





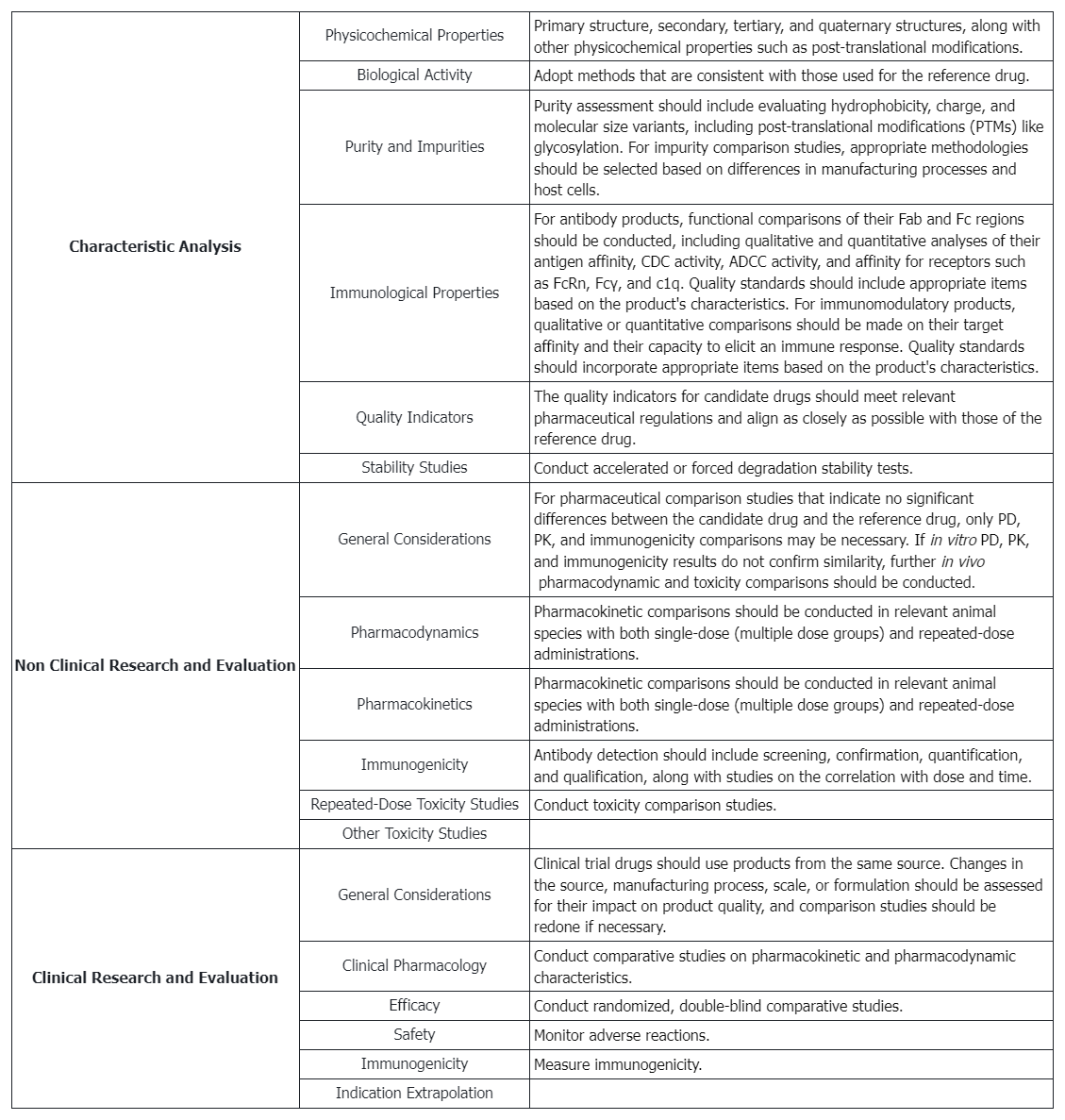
Biosimilar Assessment Service
Biosimilars are biological products that are highly similar to an approved original biological drug (reference or brand drug) without any clinically meaningful differences in quality, safety, or efficacy compared to those approved. Biosimilars typically enter the market after the patent protection period of the original drug has expired, providing a more cost-effective treatment option.
Several biosimilars have already been approved and are available on the market, primarily in expensive biological therapy areas such as oncology, immunology, and hematology. Below are some examples of approved biosimilars:
1. Adalimumab Biosimilar
Used for treating rheumatoid arthritis (RA), psoriasis, and inflammatory bowel disease (IBD).
2. Infliximab Biosimilar
Used for treating RA, Crohn's disease (CD), and ulcerative colitis (UC).
3. Filgrastim Biosimilar
Used for reducing the risk of febrile neutropenia (FN).
4. Epoetin Biosimilar
Used for treating anemia.
5. Somatropin Biosimilar
Used for treating growth hormone deficiency.
6. Glargine Biosimilar
Used for treating diabetes.
7. Rituximab Biosimilar
Used for treating non-Hodgkin lymphoma and chronic lymphocytic leukemia.
8. Bevacizumab Biosimilar
Used for treating certain types of cancer, such as metastatic colorectal cancer, non-small cell lung cancer (NSCLC), and renal cell carcinoma.
9. Trastuzumab Biosimilar
Used for treating HER2-positive breast cancer and gastric cancer.
These biosimilars usually contain the same active ingredients as the original drugs and are highly similar in terms of efficacy and safety. However, their development and approval process must meet strict regulatory requirements to ensure safety and efficacy for patients.
Advantages and Limitations of Biosimilars as Therapeutic Drugs
Strategies to Overcome Limitations Associated with Biosimilars
1. Strengthening Regulatory Frameworks
Ensure clear regulations and guidelines to the development, approval, and monitoring of biosimilars after they entering the market.
2. Increasing R&D Efficiency
Foster innovation and optimize the biosimilar development process to reduce costs and time.
3. Promoting Market Access
Accelerate the entry of biosimilars into the market by implementing policies and legislation to reduce patent obstacles set by original drug manufacturers.
4. Enhancing Interchangeability Guidance
Establish clear rules and guidelines that define the conditions under which biosimilars can be used as substitutes for original drugs.
5. Strengthening International Cooperation
Promote regulatory collaboration between countries and regions to standardize the approval processes and criteria for biosimilars.
6. Continuing Quality Control
Ensure biosimilars maintain high-quality standards throughout all stages of production and continue to monitor their safety and efficacy post-market.
7. Transparency and Data Sharing
Encourage pharmaceutical companies to share detailed data on product development and clinical trials with regulatory agencies to enhance the transparency of the approval process.
Testing Content Required by Policies and Regulations
Scientific Considerations in Demonstrating Biosimilarity to a Reference Product
When conducting biosimilarity assessments, regulatory authorities and scientific guidelines typically focus on the following factors:
1. Structural and Functional Characteristics
A comprehensive comparison of the proposed product and the reference product, including amino acid sequence, higher order structures, and post-translational modifications.
2. Manufacturing Process
Differences in the manufacturing process can affect the safety and efficacy of protein products, necessitating detailed consideration and evaluation.
3. Selection of the Reference Product
A reference product approved must be selected for direct comparison with the proposed product.
4. Animal Studies
Data from animal studies, including toxicity assessments, can support the safety evaluation of the proposed product.
5. Human Pharmacokinetic (PK) and Pharmacodynamic (PD) Studies
These studies are crucial in predicting the clinical effects of protein products, typically involving comparisons of PK and PD characteristics.
6. Clinical Immunogenicity Assessment
Evaluation of potential differences in immune response between the proposed product and the reference product in humans.
7. Clinical Studies
Additional clinical studies may be required if uncertainties about biosimilarity persist after prior evaluations.
8. Totality of the Evidence Approach
All submitted data and information will be comprehensively considered, including structural and functional characteristics, non-clinical evaluations, human PK and PD data, clinical immunogenicity data, and comparative clinical study data.
9. Post-Market Safety Monitoring
Ensuring mechanisms are in place for post-market monitoring and evaluation of safety issues.
10. Communication with Regulatory Authorities
Sponsors are encouraged to communicate early and continuously with relevant regulatory authorities to discuss product development plans and methods for providing sufficient scientific evidence.
11. Scientific Justification
Sponsors must provide scientifically sound explanations for why certain studies are unnecessary or specific study designs are appropriate.
12. Extrapolation of Data
If biosimilarity is demonstrated for certain indications, sponsors may seek approval for other indications and provide sufficient scientific evidence supports data extrapolation.
13. Product Complexity
Recognizing the complexity of protein products and considering relevant scientific issues in biosimilar development plans.
14. Risk Management
Adopting a risk-based approach to evaluate all available data and information to support the biosimilarity of the proposed product.
Biosimilars Analysis Solutions
Service Advantages
1. We provide professional and robust services, covering multiple aspects of biosimilar testing.
2. Our testing services are cost-effective, with short experimental cycles and complete, reliable reports.
3. Our laboratory is equipped with various high-resolution mass spectrometers and other advanced instruments.
Example Results
1. The First Generic Teriparatide: Structural and Biological Similarity to Its Reference Drug
Teriparatide is a synthetic anabolic peptide drug used for the treatment of osteoporosis. Recombinant Teriparatide was first approved in 2002, and subsequently, patent-free alternatives were introduced under biosimilar or hybrid regulatory applications. A study aimed to demonstrating the essential similarity between synthetic teriparatide BGW and the reference medicinal product (RMP) to ensure the development of the first biosimilar teriparatide drug. Therefore, an extensive side-by-side comparison was conducted using various state-of-the-art orthogonal methods, focusing on structure and biological activity. Nuclear magnetic resonance (NMR), ion mobility-mass spectrometry (IM-MS), UV, circular dichroism (CD), and Fourier transform infrared (FTIR) demonstrated the structural similarity between teriparatide BGW and RMP. Cell-based comparative bioassays showed that the synthetic and recombinant peptides exhibited equivalent biological activity. Teriparatide BGW, as a biosimilar, offered an available treatment option for osteoporosis patients and provided the same clinical benefits as RMP.
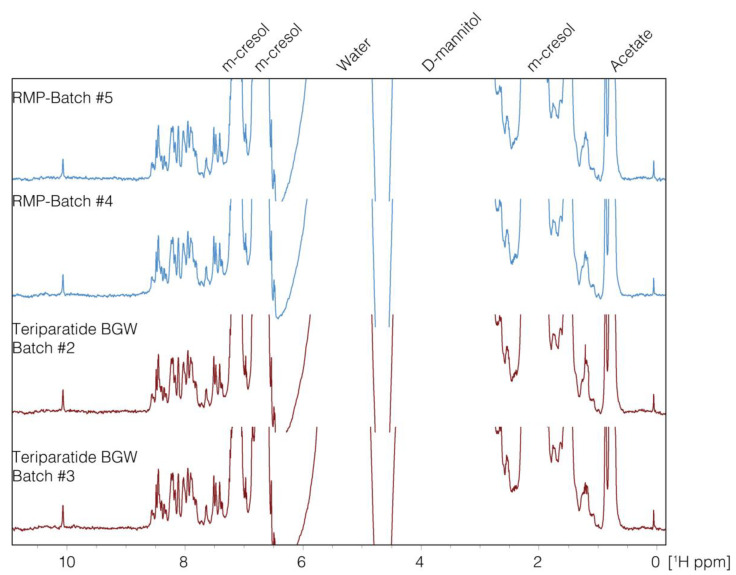
Figure 1. 1D-NMR Spectra (8.0-1.0 ppm) of Teriparatide and the Reference Drug [1]
2. Comparative Pharmacokinetic Study of Biosimilar HEC14028 and Dulaglutide in Healthy Subjects
A study aimed to evaluate the pharmacokinetics (PK), safety, and immunogenicity of biosimilar HEC14028 compared to the reference product dulaglutide in healthy male subjects. This study was a single-center, randomized, open-label, single-dose, parallel-controlled comparative phase I clinical trial, including up to 14 days of screening, 17 days of observation post-dosing, and 7 days of safety follow-up. A total of 68 healthy male subjects were randomly assigned (1:1) to the test group (HEC14028) and the reference group (dulaglutide) (single 0.75 mg subcutaneous abdominal injection). The primary objective was to evaluate the PK characteristics of HEC14028 and to compare the PK similarity between HEC14028 and dulaglutide. The primary PK endpoints were the maximum plasma concentration (Cmax) and the area under the blood concentration-time curve from zero time to estimated infinity time (AUC0-∞). The results showed that the PK of HEC14028 and dulaglutide were pharmacokinetically comparable: the 90% confidence intervals (CI) for the geometric mean ratios of Cmax and AUC0-∞ were 102.9%-122.0% and 97.1%-116.9%, respectively, both within the 80.00%-125.00% range. No grade 3 or higher treatment emergent adverse events (TEAEs), serious adverse events (SAEs), TEAEs leading to trial withdrawal, or TEAEs leading to death were reported. Both HEC14028 and dulaglutide showed good and similar safety profiles, with no increased immunogenicity observed in subjects receiving HEC14028 and dulaglutide.
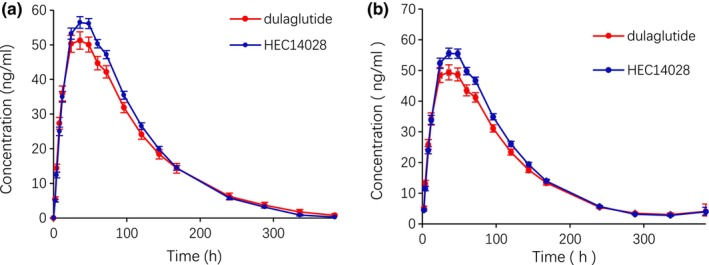
Figure 2. Mean Plasma Concentration-Time Curves after a Single Subcutaneous Injection of HEC14028 or Dulaglutide [2]
3. Characterization of PEGylation Sites in Neulasta and Mass Spectrometric Analysis of Biosimilar Candidates Using a Combined Fragmentation Strategy
In developing biosimilar products for Neulasta, it is essential to determine the intact molecular mass and confirm the precise PEGylation sites. A study employed a combination of techniques, including the post-column addition of triethylamine in reversed-phase liquid chromatography-mass spectrometry (RPLC-MS) to determine the intact molecular mass, and in-source fragmentation (ISF) along with higher-energy collision dissociation-tandem mass spectrometry (HCD-MS/MS) to identify the PEGylation sites. The results indicated that both the pegfilgrastim biosimilar candidate and Neulasta lots were singly PEGylated at the N-terminual end. Additionally, the study demonstrated that the combination of ISF and HCD-MS/MS could be used to identify PEGylation sites in di-PEGylated variants of pegfilgrastim. The diPEGylated variant had modification sites at the N-terminal and a lysine at position 35 in the protein sequence.
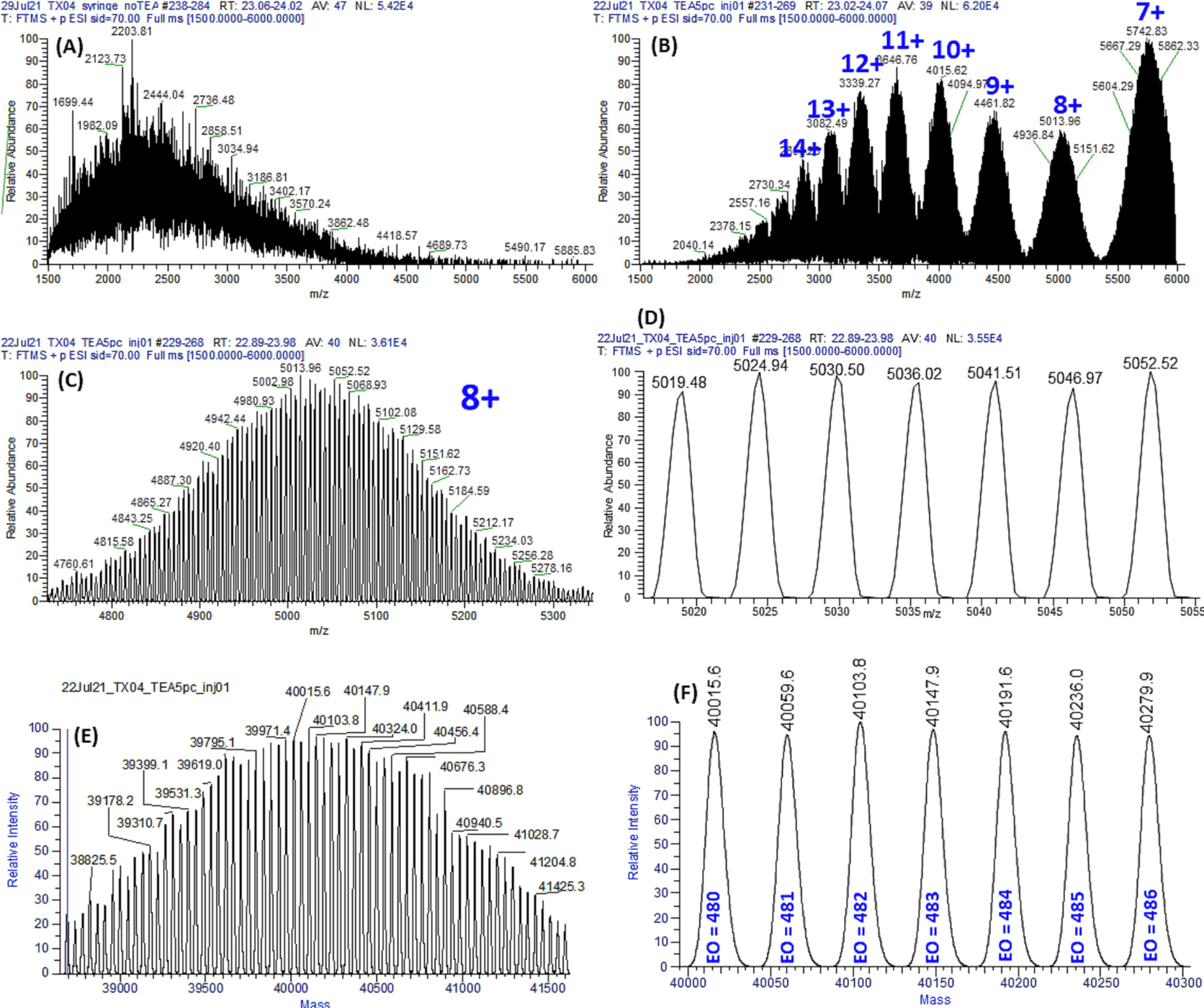
Figure 3. Mass Spectrum of the Pegfilgrastim Biosimilar Candidate [3]
4. Comparison of Physicochemical and Structural Characterization Studies Demonstrating High Biosimilarity Between BGL-ASP and Reference Insulin Aspart
Biosimilar insulin analogues are improving global market access for diabetes patients. Establishing scientific biosimilarity is crucial for this change in medical landscape. BGL-ASP is a biosimilar insulin aspart developed by BioGenomics Limited, India. BioGenomics adopted a stepwise approach to gather all necessary evidence to establish similarity with the reference product. Insulin aspart is a recombinant fast-acting human insulin analogue used to treat type-1 and type-2 diabetes mellitus. The substitution of proline with aspartic acid at position B28 reduces the tendency to form hexamers, thereby increasing absorption rates upon subcutaneous administration compared to native insulin. To determine the safety and efficacy of BGL-ASP, the critical quality attributes (CQAs) of BGL-ASP were identified based on their impact created on biological activity, PK/PD, immunogenicity, and safety. The CQAs of insulin aspart related to product structure, purity, and functionality, characterized using advanced orthogonal analytical tools. The primary, secondary, tertiary, and quaternary structures were found to be highly similar for BGL-ASP and reference product. High performance liquid chromatography (HPLC) analyses determined product-related impurities and assay content of insulin aspart, which were similar to the reference insulin aspart from the US, EU, and India. The safety, efficacy, and immunogenicity of BGL-ASP were also found to be comparable to the reference product, confirmed through clinical trials conducted in accordance with International Council for Harmonisation of Technical Requirements of Pharmaceuticals for Human Use (ICH) and European Medicines Agency (EMA) guidelines. The data in this study indicated a high degree of similarity in structural, physicochemical, and biological characteristics between reference insulin aspart and BGL-ASP, confirming its safety and efficacy as a potential cost-effective therapeutic alternative for diabetes mellitus treatment.
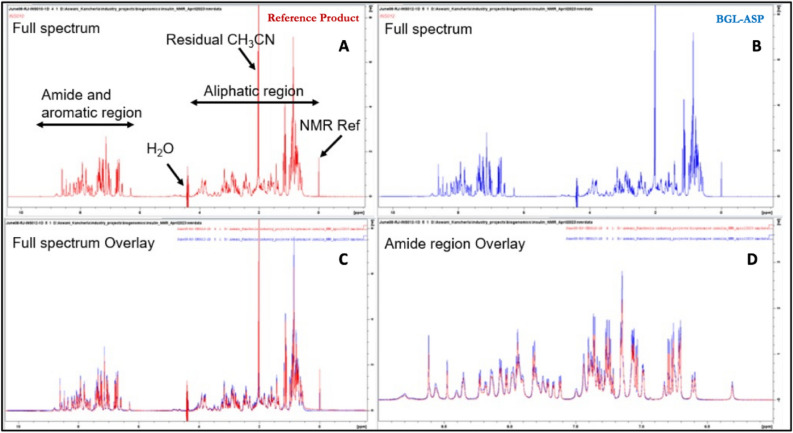
Figure 4. Comparison of 1D Proton Spectra of BGL-ASP and the Reference Product [4]
References
[1] Fernández-Carneado J, Vallès-Miret M, Arrastia-Casado S, Almazán-Moga A, Macias MJ, Martin-Malpartida P, Vilaseca M, Díaz-Lobo M, Vazquez M, Sanahuja RM, Gambús G, Ponsati B. First Generic Teriparatide: Structural and Biological Sameness to Its Reference Medicinal Product. Pharmaceutics. 2024 Apr 13;16(4):537. doi: 10.3390/pharmaceutics16040537. PMID: 38675198; PMCID: PMC11054030.
[2] Gao X, Di Y, Lv Y, Luan Y, Xiong Y, Xu Y, Li Y, Guo L, Li X, Deng L, Zhuang Y, Hou J. A pharmacokinetic study comparing the biosimilar HEC14028 and Dulaglutide in Healthy Subjects. Clin Transl Sci. 2024 Apr;17(4):e13775. doi: 10.1111/cts.13775. PMID: 38651744; PMCID: PMC11036873.
[3] Rauniyar N, Togle AJ, Ronci RA, Reyes D, Han X. Characterization of PEGylation sites in Neulasta and a biosimilar candidate with a combined fragmentation strategy in mass spectrometry analysis. J Mass Spectrom. 2024 Apr;59(4):e5017. doi: 10.1002/jms.5017. PMID: 38517094.
[4] Ghade NS, Thappa DK, Lona J, Krishnan AR, Sonar SM. Comparative physicochemical and structural characterisation studies establish high biosimilarity between BGL-ASP and reference insulin aspart. Sci Rep. 2024 Feb 20;14(1):4224. doi: 10.1038/s41598-024-54819-x. PMID: 38378730; PMCID: PMC10879530.
How to order?

