





Generic Peptide Pharmaceuticals Assessment Service
Generic peptide drugs are products that replicate existing peptide drugs. These drugs are typically composed of amino acids linked by peptide bonds and are used to treat a variety of diseases, including cancer, diabetes, and immune disorders. Developing these generics requires comprehensive research into synthesis processes, formulation technologies, quality control, and bioequivalence to ensure they match the quality and efficacy of the original drugs.
Some representative generic peptide drugs include:
1. Glatiramer Acetate
A treatment for multiple sclerosis.
2. Liraglutide
A glucagon-like peptide-1 (GLP-1) receptor agonist used to treat type 2 diabetes.
3. Semaglutide
Another GLP-1 receptor agonist for type 2 diabetes, also studied for weight loss.
4. Albiglutide
A GLP-1 receptor agonist for type 2 diabetes.
5. Somatuline
Used for treating acromegaly and certain neuroendocrine tumors.
6. Teriparatide
A medication for severe osteoporosis.
7. Abatacept
Treats autoimmune diseases such as rheumatoid arthritis.
Advantages and Limitations of Generic Peptide Drugs as Therapeutic Drugs
1. Advantages
(1) High Activity and Selectivity: Peptide drugs often have higher activity and greater selectivity, advantageous for treating complex diseases.
(2) Fewer Side Effects: As peptides are composed of amino acids, their metabolites are also amino acids, resulting in fewer side effects.
(3) High Stability and Purity: Peptide drugs have better stability and purity compared to protein drugs, with lower production costs and minimal immunogenicity.
(4) Quality Control: The quality control level of peptide drugs can approach that of traditional small-molecule drugs, ensuring reliability and consistency in treatment.
(5) Wide Therapeutic Range: Peptide drugs can treat various diseases, including cancer, diabetes, and obesity.
(6) Innovative Potential: Peptide drugs can be chemically modified to improve the affinity, solubility, and pharmacokinetic (PK) properties of drug candidates.
2. Limitations
(1) Stability Issues: Peptide drugs are less stable and may present special safety concerns like immunogenicity, requiring stringent storage and transportation conditions.
(2) Production Costs: The higher production costs compared to small-molecule drugs can limit their widespread use.
(3) Technical Challenges: The synthesis and quality control of peptide drugs are complex, especially for long peptides in large-scale production.
(4) Regulatory Challenges: Incomplete technical requirements and regulatory gaps exist for peptide drugs.
(5) Patent and Market Access: Even after patent expiration, generic peptide drugs face challenges in market entry and patent disputes.
(6) Raw Material Accessibility: Accessibility of high-quality peptide Active Pharmaceutical Ingredients (APIs) limits peptide drug development.
Strategies to Overcome Limitations Associated with Generic Peptide Drugs
1. Improve Stability and Half-Life
Enhance peptide stability through N- or C-terminal modifications, such as methylation or acetylation. Use D-amino acids and non-natural amino acids to reduce protease affinity. Cyclize or bicylize peptides to increase rigidity. Increase molecular weight with the addition of fatty chains or PEGylation to improve plasma half-life.
2. Improve Bioavailability
Use bioavailable peptides for oral administration, enhancing oral absorption with penetration enhancers. Develop new delivery systems like needle-free injections, microneedles, and pulmonary routes to reduce dependency on traditional injections.
3. Enhance Solubility and Affinity
Improve solubility and target affinity through main-chain and side-chain modifications to stabilize secondary structures.
4. Rational Design
Establish structure-activity relationships (SAR) from known peptide crystal structures to enhance physicochemical properties.
5. Genetic Code Expansion
Incorporate non-canonical amino acids (ncAAs) with novel properties into peptides using genetic code expansion to increase functionality.
6. PEGylation
Increase effective molecular weight with PEGylation to reduce renal clearance and enhance half-life.
7. Lipid and Protein Conjugation
Conjugate with fatty acids or larger proteins to extend plasma circulation time.
8. Develop Multifunctional Peptides
Design peptides with multiple biological activities for personalized and comprehensive treatment plans.
9. Improve Production Technology
Use solid-phase peptide synthesis (SPPS) and automated methods to enhance synthesis efficiency and purity.
10. Purification and Analysis
Employ techniques like reverse-phase high-performance liquid chromatography (RP-HPLC) and capillary electrophoresis (CE) for peptide separation, purification, and quality control.
Testing Content Required by Policies and Regulations
The quality standards of peptide drugs cover various aspects such as chemical, physical, and biological properties as well as quality control measures during the production process. The specific quality requirements are as follows:
1. Chemical and Physical Characteristics
(1) Defined Chemical Structure: The amino acid sequence, molecular weight, stereochemistry, and any modifications (such as PEGylation) of peptide drugs must be clearly defined.
(2) Purity and Impurity Control: The purity of peptide drugs must meet specified standards, and impurity levels (including peptide-related and non-peptide-related impurities) must be controlled within safe limits.
(3) Stability: Peptide drugs must remain stable under expected storage and usage conditions. Stability studies should include both accelerated and long-term testing.
2. Production Process
(1)Compliance: The production of peptide drugs must adhere to Current Good Manufacturing Practices.
(2) Production Consistency: The production process should ensure consistency between batches, including synthesis, purification, and formulation steps.
(3) Validation and Confirmation: Key parameters and control measures of the production process must be validated and confirmed.
3. Quality Control Testing
(1) Comprehensive Testing Plan: A detailed quality control testing plan should include tests for raw materials, intermediates, and final products.
(2) Validation of Test Methods: All quality control test methods must be validated to ensure the accuracy and reliability of results.
4. Bioequivalence and Preclinical Data
(1) Bioequivalence: For generic drugs, bioequivalence to the reference drug must be demonstrated.
(2) Preclinical Data: For new drugs, preclinical data on pharmacology, toxicology, and PK must be provided.
5. Labeling and Instructions
Drug labels and instructions should include all relevant safety, efficacy, and usage information.
6. Immunogenicity Assessment
For peptide drugs with potential immunogenicity, an immunogenicity risk assessment must be conducted, and clinical immunogenicity evaluation should be performed if necessary.
7. Drug Interactions
Potential drug interactions, particularly effects on cytochrome P450 (CYP) enzymes and drug transport proteins, should be evaluated.
8. Regulatory Pathways
Appropriate regulatory pathways should be selected based on the drug's characteristics and development stage, such as Abbreviated New Drug Application (ANDA) or New Drug Application (NDA).
9. Combination Products
For combination products containing peptide or protein components, quality assessment may involve collaboration among multiple sub-offices within Ofice of Pharmaceutical Quality (OPQ).
10. Change Management
Any changes in the production process must undergo appropriate change management.
ANDAs for Certain Highly Purified Synthetic Peptide Drug Products That Refer to Listed Drugs of rDNA Origin
1. Introduction
This guidance provides recommendations for evaluating whether an ANDA submission is appropriate for a synthetic peptide that references any of the following five previously approved peptide drug products of rDNA origin: glucagon, liraglutide, nesiritide, teriparatide, and teduglutide. An ANDA should demonstrate that the synthetic peptide has the same active ingredient as the referenced drug of rDNA origin and is comparable in terms of purity, safety, and efficacy.
2. Background
According to the Federal Food, Drug, and Cosmetic Act (FD&C Act), any α-amino acid polymer containing 40 or fewer amino acids is defined as a peptide, not a protein. ANDA approval requires demonstrating that the proposed generic drug is the same as the reference listed drug (RLD) in terms of active ingredients, conditions of use, dosage form, route of administration, strength, and (with certain allowable differences) labeling and is bioequivalent to it.
3. Scientific Considerations for ANDA Submission
(1) Active Ingredient Sameness: Determined through physicochemical characterization and biological evaluations (such as clinical PK and/or pharmacodynamic (PD) studies).
(2) Impurities: During ANDA review, the differences in the types and quantities of impurities in the proposed generic drug compared to the RLD are considered. Synthetic peptides should not contain impurity levels higher than those in the RLD, and any impurities, including new impurities, should be reasonably justified to ensure the generic drug does not pose greater safety risks, including immunogenicity risks.
Clinical Pharmacology Considerations for Peptide Drug Products
1. Introduction
This guidance provides recommendations on clinical pharmacology considerations in the development of peptide drug products, including the effects of hepatic impairment, drug-drug interactions (DDIs), QT interval prolongation risk, and immunogenicity risk on the PK, safety, and efficacy of peptide drug products.
2. Definition of Peptide Drug Products
Peptides are defined as polymers composed of 40 or fewer amino acids. If a peptide meets the definition of a drug and does not meet the statutory definition of a “biological product” or “device,” it is regulated as a drug under the FD&C Act.
3. Clinical Pharmacology Assessment
(1) Immunogenicity Risk Assessment: Most peptide drug products have the potential for immunogenicity, requiring an assessment of the immunogenicity risk for all peptide drug products.
(2) Effects of Hepatic Impairment: Although peptide drug products are typically extensively metabolized by proteases and peptidases, it may be important to assess the impact of hepatic impairment on the PK of peptide drug products in certain cases.
(3) Drug Interaction Evaluation: Peptide drug products are generally not metabolized by CYP enzymes and are therefore not affected by CYP enzyme inhibitors or inducers. However, certain structurally modified peptides (e.g., cyclic peptides) may be subject to CYP enzyme-mediated metabolism and transporter-mediated disposition.
(4) QT Interval Prolongation Characterization: QT interval studies are clinical trials conducted during drug development to assess a new drug's effect on the QT interval of the heart. Peptides composed of naturally occurring amino acids generally have a low likelihood of direct ion channel interactions, usually not requiring thorough QT studies unless there are mechanistic considerations or preclinical data suggesting potential arrhythmia risk.
Analysis Solutions
Service Advantages
1. Professional and Robust Services Covering a Wide Range of Recombinant Protein Drug Testing
2. High Cost-Effectiveness, Short Testing Cycles, and Reliable, Comprehensive Test Reports
3. Equipped with a Variety of High-Resolution MS and other Major Equipment
Sample Results
1. Characterization of Low-Level D-Amino Acid Isomer Impurities of Semaglutide Using Liquid Chromatography-High Resolution Tandem Mass Spectrometry
According to guidelines for ANDAs for Certain Highly Purified Synthetic Peptide Drug Products That Refer to Listed Drugs of rDNA Origin Guidance for Industry, a synthetic Semaglutide that is intended to be a “generic” of the approved rDNA origin Semaglutide is under exploring. Therefore, for Semaglutide covered by this guideline, it is necessary to identify each peptide-related impurity present at 0.10 % or higher of the drug substance. Characterizing low-level D-amino acid (D-form) isomeric impurities is always the most challenging. Impurities were separated using reverse-phase ultra performance liquid chromatography (RP-UPLC and the molecular weight of impurities present in two formulations was determined using high resolution mass spectrometry (HRMS). After targeted off-line collection of D-isomers, samples were lyophilized, hydrolyzed with deuterated hydrochloric acid (D-HCl), and subjected to low-level D/L-form transfer inhibition substrates, chiral derivatization, and RP-UPLC tandem mass spectrometry analysis, compared with standards. This study reports an accurate, direct characterization method with low detection limits for low-level D-Ser8, D-His1, and D-Asp9 impurities in Semaglutide preparations. The developed UPLC-HRMS method takes a valuable step forward in detecting trace D-isomers in Semaglutide and other peptide products.
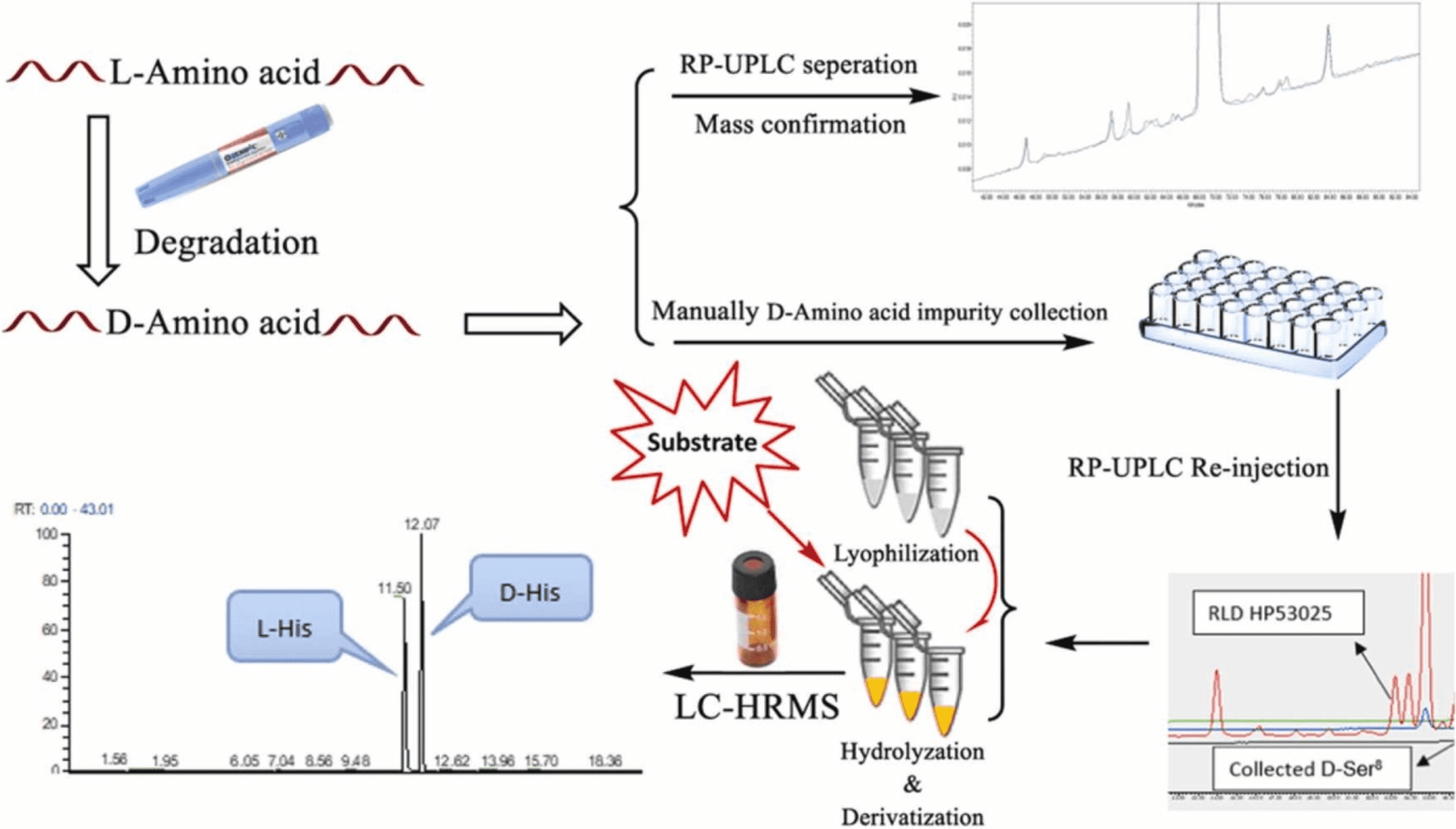
Figure 1. Separation of Impurities RP-UPLC and Determination of Molecular Weight of Impurities Present in Two Formulations Using HRMS [1]
2. Demonstration the Equivalence of Generic Glatiramer Acetate (Glatopa™)
Glatiramer acetate (GA) has been available under the brand name Copaxone® for nearly twenty years. Recently, the first generic GA, Glatopa™, was approved as a complete replacement for all approved indications for Copaxone 20 mg. Glatopa is also the first “AP-rated” interchangeable generic medicine approved for the treatment of patients with MS. Glatiramer acetate is a complex peptide mixture, presenting challenges not typically encountered in drug development. Despite its complexity and lack of clinical data, approval was achieved through an ANDA, demonstrating equivalence to Copaxone based on four criteria: starting materials and basic chemistry; structural signatures for polymerization, depolymerization, and purification; physicochemical properties; and biological and immunological properties. This article describes the rigorous, comprehensive scientific approach used to establish the equivalence between Glatopa and Copaxone and presents key representative data from the comprehensive physicochemical (structural) and biological (functional) assays conducted.
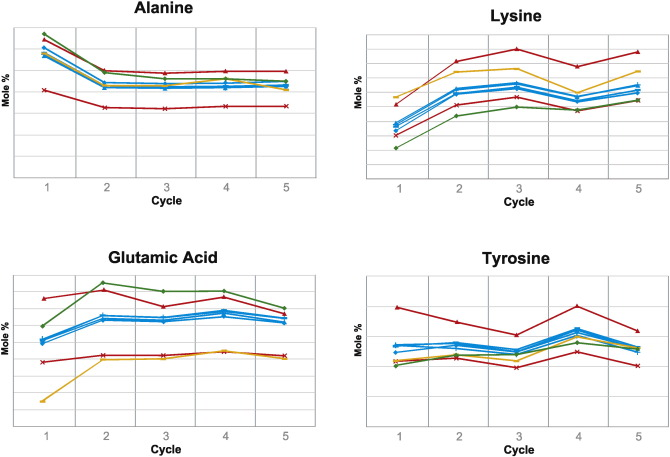
Figure 2. Relative Amino Acid Levels at the N-Terminus of Glatiramer Acetate for the First Five Cycles of N-Terminal Analysis by Edman Degradation [2]
3. Comparison of Biological Impacts of Glatiramer Acetate and Its Generic
For decades, policies on generic drugs have aimed to provide patients with affordable, safe, and effective medications while encouraging the development of new therapies. For physicians and regulators, balancing this has become increasingly challenging as biologics and non-biological complex drugs (NBCDs) like glatiramer acetate show significant efficacy, making their generics harder to evaluate. We aimed to develop computational methods to compare the biological impacts of brand-name drugs and generics using transcriptome profiling, characterizing biological impact differences. We integrated multiple computational methods to determine if differentially expressed genes resulted from random variation or pointed to consistent biological impact differences between the branded and generic drugs. These methods analyzed gene expression data from mouse splenocytes exposed to brand-name or generic glatiramer acetate. Computational methods provided substantial evidence that brand-name glatiramer acetate exhibited more consistent biological impacts between batches compared to the generic, with significant effects on regulatory T cells and myeloid cells. In summary, we developed a computational pipeline integrating multiple methods to innovatively compare the two drugs. This pipeline and specific findings distinguishing brand-name glatiramer acetate from its generic can help physicians and regulators take appropriate measures to ensure safety and efficacy.
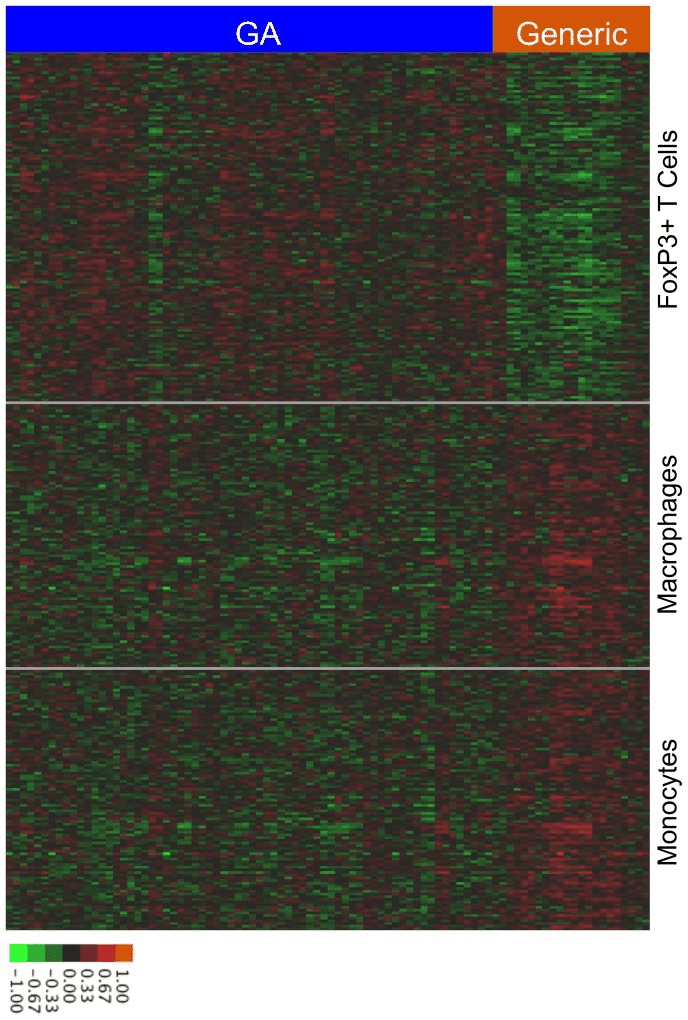
Figure 3. Cell Type-Specific Differences in the Biological Impacts of GA and Its Generic [3]
References
[1] Zhang B, Xu W, Yin C, Tang Y. Characterization of low-level D-amino acid isomeric impurities of Semaglutide using liquid chromatography-high resolution tandem mass spectrometry. J Pharm Biomed Anal. 2023 Feb 5;224:115164. doi: 10.1016/j.jpba.2022.115164. Epub 2022 Nov 16. PMID: 36462248.
[2] Anderson J, Bell C, Bishop J, Capila I, Ganguly T, Glajch J, Iyer M, Kaundinya G, Lansing J, Pradines J, Prescott J, Cohen BA, Kantor D, Sachleben R. Demonstration of equivalence of a generic glatiramer acetate (Glatopa™). J Neurol Sci. 2015 Dec 15;359(1-2):24-34. doi: 10.1016/j.jns.2015.10.007. Epub 2015 Oct 8. PMID: 26671082.
[3] Towfic F, Funt JM, Fowler KD, Bakshi S, Blaugrund E, Artyomov MN, Hayden MR, Ladkani D, Schwartz R, Zeskind B. Comparing the biological impact of glatiramer acetate with the biological impact of a generic. PLoS One. 2014 Jan 8;9(1):e83757. doi: 10.1371/journal.pone.0083757. PMID: 24421904; PMCID: PMC3885444.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

