





Avoid Inaccurate Results With These Advanced Protein Identification Methods
In proteomics research, data bias is a major factor contributing to inaccurate and inconsistent protein identification results. Variability introduced at any stage-ranging from sample preparation to experimental execution and data analysis-can compromise data integrity and hinder reproducibility. Without proper control, these biases may obscure biological interpretations and mislead scientific conclusions. This paper provides a systematic analysis of common sources of bias in protein identification and outlines state-of-the-art strategies to mitigate these issues, ensuring the accuracy and reliability of experimental data.
Sources of Data Bias in Protein Identification
The primary contributors to data bias in protein identification include:
1. Sample-Related Biases
Variability in sample preparation, protein degradation, and selective loss of low-abundance proteins can skew protein identification outcomes.
2. Instrumental Biases
Mass spectrometry signal drift and batch effects introduce inconsistencies between experimental runs.
3. Data Analysis Biases
Errors in database searches, incorrect peptide-to-protein mapping, and false-positive identifications affect the reliability of results.
4. Biological Variability
Intrinsic sample heterogeneity and biological fluctuations introduce additional complexity, making it challenging to distinguish technical artifacts from true biological differences.
Addressing these biases is essential for obtaining reproducible and biologically meaningful results in proteomics studies. Implementing standardized protocols, rigorous quality control measures, and advanced data processing algorithms can significantly enhance the precision of protein identification.
Advanced Strategies for Correcting Data Bias in Protein Identification
1. Optimizing Sample Processing to Minimize Pre-Analytical Bias
(1) High-Fidelity Protein Extraction and Processing
①Selecting appropriate lysis buffers and incorporating protease/phosphatase inhibitors to prevent protein degradation.
②Enriching low-abundance proteins by depleting high-abundance proteins using specialized reagents (e.g., plasma protein depletion columns).
③Standardizing protein quantification (e.g., BCA, Bradford) and enzymatic digestion (dual-enzyme Trypsin + LysC) to enhance digestion reproducibility.
(2) Controlling Batch Effects in Sample Preparation
①Employing internal standard proteins (e.g., SILAC, iTRAQ/TMT labeling) for data normalization.
②Utilizing automated proteomics platforms to reduce operator-induced variability.
2. Advanced Mass Spectrometry Calibration Techniques
(1) Internal Calibration for Mass Accuracy
①Implementing lock mass correction to compensate for instrumental drift and enhance m/z precision.
②Using pooled sample strategies to mitigate batch-to-batch signal variation.
(2) Minimizing Quantitative Errors in Mass Spectrometry
①Adopting data-independent acquisition (DIA) to eliminate stochastic peptide sampling biases.
②Leveraging high-resolution mass analyzers (Orbitrap, FT-ICR) to reduce signal noise and improve quantitative reliability.
3. Cutting-Edge Data Analysis Approaches to Enhance Protein Identification
(1) AI-Driven Bias Mitigation in Proteomics
①Machine learning algorithms (e.g., Percolator) refine database searches by optimizing false discovery rates (FDR).
②Deep learning-based tools (e.g., DeepNovo) improve de novo sequencing and correct mass spectral peak mismatches.
(2) Dynamic FDR Control for Increased Identification Confidence
①Conventional FDR methods (e.g., PeptideProphet) often introduce false positives in low-abundance proteins.
②Local-FDR models dynamically adjust thresholds based on abundance levels, enhancing confidence in low-abundance protein identifications.
(3) Integrated Multi-Database Search Strategies
①Combining multiple protein databases (Uniprot, NCBI RefSeq, SwissProt) to increase identification accuracy.
②Employing open-search algorithms (e.g., MetaMorpheus) to improve PTM detection and reduce undetected peptide loss.
4. Statistical and Normalization Models for Batch Effect Reduction
(1) Removing Batch Effects via Statistical Correction
①Applying Bayesian-based Combat algorithms to correct technical batch variations.
②Utilizing LOESS normalization to adjust for inter-sample signal intensity discrepancies.
(2) Normalization Techniques for Accurate Quantification
①Z-score normalization for datasets with high variability in protein expression.
②Variance Stabilization Normalization (VSN) for stabilizing low-abundance protein quantification.
5. Experimental Validation to Ensure Data Robustness
(1) Mass Spectrometry-Based Validation
①Conducting PRM or SRM quantification to validate key protein identifications.
②Enhancing data consistency with iTRAQ/TMT-based quantification combined with DIA.
(2) Immunological and Functional Validation
①Using Western blot and Co-IP coupled with MS to confirm protein interactions.
②Employing CRISPR/Cas9 knockout models and single-cell proteomics (SCoPE-MS) to validate biological function.
Conclusion: Strategies to Minimize Data Bias in Protein Identification
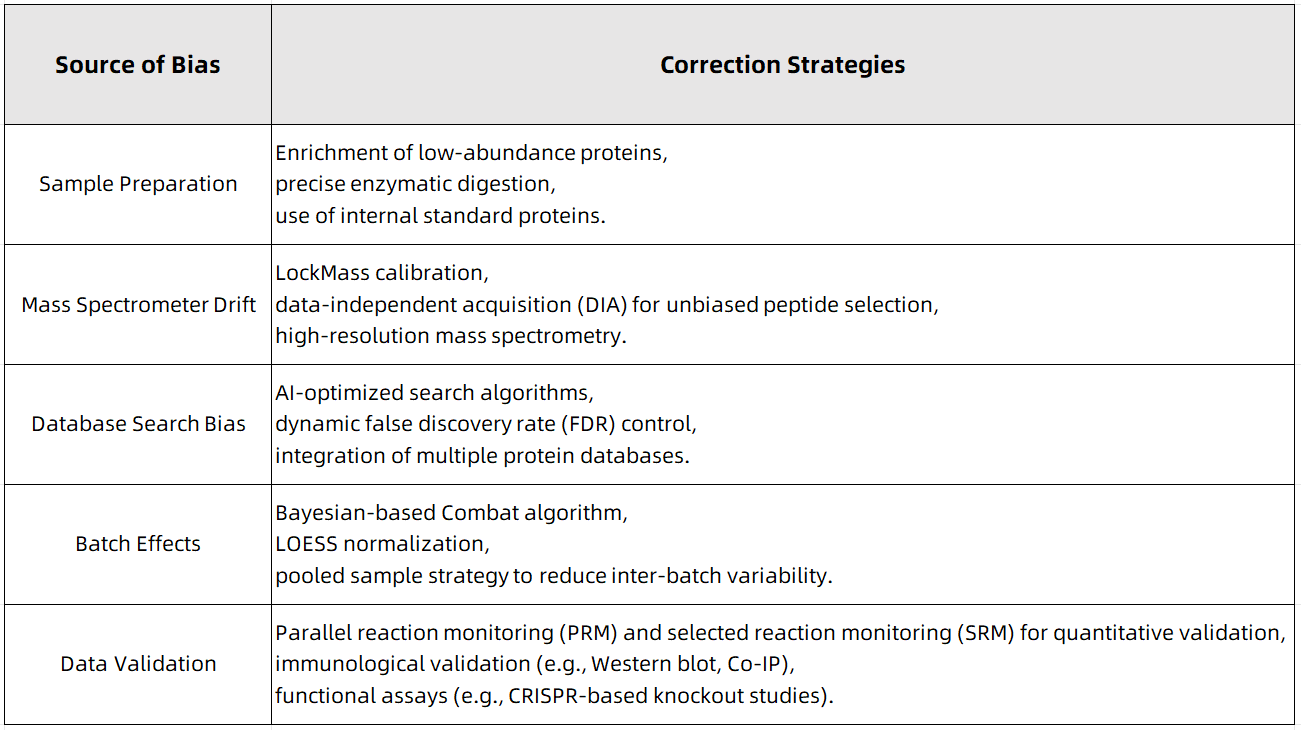
Figure 1
Achieving high-precision protein identification requires a comprehensive approach, including rigorous experimental workflow optimization, advanced computational data analysis, systematic bias correction, and multi-modal experimental validation. By applying these bias correction techniques, researchers can significantly enhance data reproducibility, minimize false discoveries, and improve confidence in protein identification results.
At MtoZ Biolabs, we leverage high-resolution mass spectrometry platforms to provide accurate and reproducible protein identification services. With extensive experience in proteomics research, we are dedicated to delivering customized, high-quality solutions for your scientific needs.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

