





How to Perform Protein Sequence Analysis?
Protein sequence analysis involves examining the amino acid sequences of proteins to elucidate their structures and functions. This analytical approach transcends basic structural characterization, providing critical insights into protein functionality, molecular interactions, disease mechanisms, and therapeutic targets. Proteins exhibit remarkable diversity, with each protein's unique three-dimensional structure directly linked to its specific function, determined by its amino acid sequence. Understanding and determining these sequences is fundamental to studying protein functionality and expression. For example, sickle cell anemia arises from a single amino acid substitution, where glutamic acid at position six of the β-globin chain is replaced by valine, forming aberrant sickle hemoglobin that replaces normal hemoglobin. Protein sequence analysis enables researchers to identify the amino acid composition of unknown proteins, detect post-translational modifications, and explore the relationship between structure and function. The sequence and N/C terminal of proteins are critical for protein synthesis and overexpression: (1) During protein synthesis, the half-life of a protein is determined by specific amino acids at the N-terminal of the polypeptide chain, which play a regulatory role in protein stability and degradation. Additionally, the N-terminal often undergoes various post-translational modifications, such as acetylation of histone N-terminal tails, which significantly impact protein function and structural stability. (2) During protein expression, external peptidases may act on the protein, potentially causing truncation of amino acids at the N/C-terminal, thereby affecting the integrity and function of the expressed protein.
Protein sequence analysis has wide-ranging applications in biomedicine, including disease diagnosis, targeted therapy, antibody drug development, and vaccine design. In drug development, it ensures the purity and stability of recombinant protein drugs, verifying their safety and efficacy. In disease research, sequence analysis of mutated or aberrantly expressed proteins uncovers molecular mechanisms of disease progression, providing vital insights for precision medicine.
Protein Sequence Analysis Methods
1. Edman Degradation
The Edman degradation method is a standard technique for analyzing the N-terminal sequence of proteins or peptides. It is widely used in biologics development, where determining the N-terminal sequence of up to 15 amino acids is a mandatory requirement for regulatory submissions. This method is also a common quality control procedure for approved biologics during annual evaluations.
The process involves reacting the protein or peptide with phenyl isothiocyanate under mildly alkaline conditions, followed by acid treatment. This selectively cleaves the N-terminal residue from the polypeptide chain as a phenylthiohydantoin (PTH) derivative, which is then analyzed, typically by liquid chromatography. The remaining peptide undergoes repeated cycles of this reaction to sequentially determine additional residues. The method is also referred to as the PTH method, due to its derivatization chemistry.
The Edman degradation method can sequence up to 50–60 residues consecutively. Key advantages include:
(1) precise identification of N-terminal amino acids, with the ability to distinguish between closely related residues such as isoleucine and leucine or glutamine and lysine;
(2) high sensitivity;
(3) direct analysis without dependence on databases, unlike mass spectrometry.
Nonetheless, this method has certain limitations: it is low-throughput, identifying only one residue per cycle; it cannot sequence directly if the N-terminal is blocked or modified, requiring prior removal of such modifications; and it is unable to analyze cysteine residues that are part of disulfide bonds without prior reduction and alkylation.
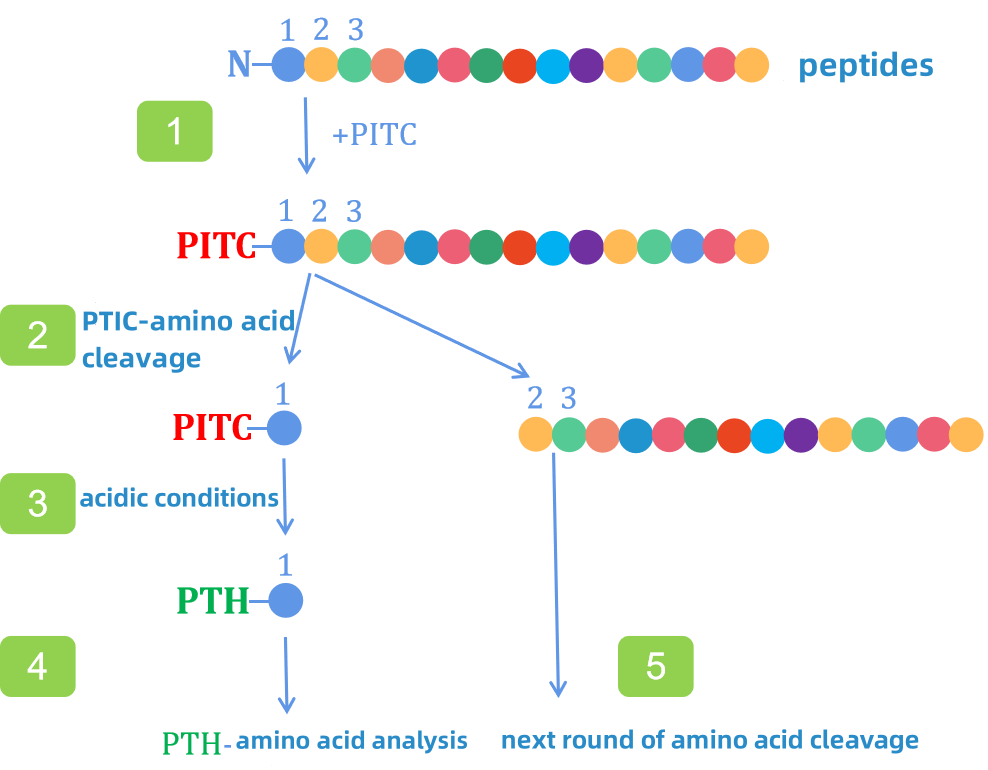
Figure 1. Edman Degradation Process
2. Mass Spectrometry
Mass spectrometry (MS), particularly tandem mass spectrometry (MS/MS), is a widely employed technique for protein amino acid sequence analysis, distinguished by its high throughput and efficiency. Proteins are enzymatically digested into peptide fragments of varying lengths (commonly 5–30 amino acids), which are analyzed by LC-MS/MS. The resulting data is matched against theoretical sequence databases, allowing for the identification of the N-terminal sequence. Notably, mass spectrometry can detect modifications or blockages at the protein N-terminus.
In cases where the polypeptide chain is cleaved into only two or three fragments, deducing their amino acid sequences and reconstructing their original order is relatively straightforward, provided the N- and C-terminal residues of the original chain are known. However, when terminal residues overlap with cleavage sites, sequence reconstruction may be ambiguous. More commonly, peptide fragmentation results in a higher number of fragments, which complicates determining the order of intermediate sequences. To overcome this, polypeptide samples are cleaved using two or more enzymatic methods with distinct cleavage specificities. This generates multiple sets of peptide fragments with overlapping sequences, known as overlapping peptides.
Overlapping peptides facilitate the accurate reconstruction of the original polypeptide chain's sequence by determining the correct order of fragments. Additionally, multiple fragment sets enable cross-validation to ensure the accuracy of individual sequence determinations. This approach constitutes the process of full-length amino acid sequence reconstruction. When reference databases are available, MS can verify the consistency between the analyzed protein and the theoretical sequence. For newly isolated proteins lacking theoretical references, MS can determine the primary structure through a process called de novo sequencing. Due to its simplicity, speed, and high throughput, mass spectrometry has become the preferred method for full-length protein sequencing.

Figure 2.
MtoZ Biolabs leverages extensive expertise in proteomics and advanced technical platforms to deliver precise and efficient protein sequence analysis services for research institutions, pharmaceutical companies, and biotechnology firms. Our team of seasoned professionals customizes analytical strategies to meet the unique requirements of each project, ensuring reliable and high-quality data. From basic research to drug development and clinical diagnostics, MtoZ Biolabs offers comprehensive support and innovative solutions, empowering the transformation of scientific discoveries into practical applications. Partnering with MtoZ Biolabs means choosing a trusted, efficient, and professional collaborator for protein sequence analysis.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

