





Applications of Single-Cell Sequencing
Humans are highly organized systems comprised of approximately 3.72×1013 cells of various types, which maintain proper organ function and normal cellular homeostasis. Despite significant and revolutionary discoveries in the field of cell biology, the heterogeneity of cells still requires further investigation. Single-cell RNA sequencing (scRNA-seq) has become the latest method for revealing the heterogeneity and complexity of RNA transcripts within individual cells, as well as for identifying different cell types and functional compositions within highly organized tissues/organs/organisms. Since its inception in 2009, studies based on scRNA-seq have provided a wealth of information across different fields, leading to exciting findings about the composition and interactions of cells in humans, model animals, and plants.
As scRNA-seq advances, the number of cells it can analyze has increased, while the cost has exponentially decreased, and the number of published papers has continuously risen. In the past decade, the technology has evolved through more complex, accurate, and high-throughput analyses.
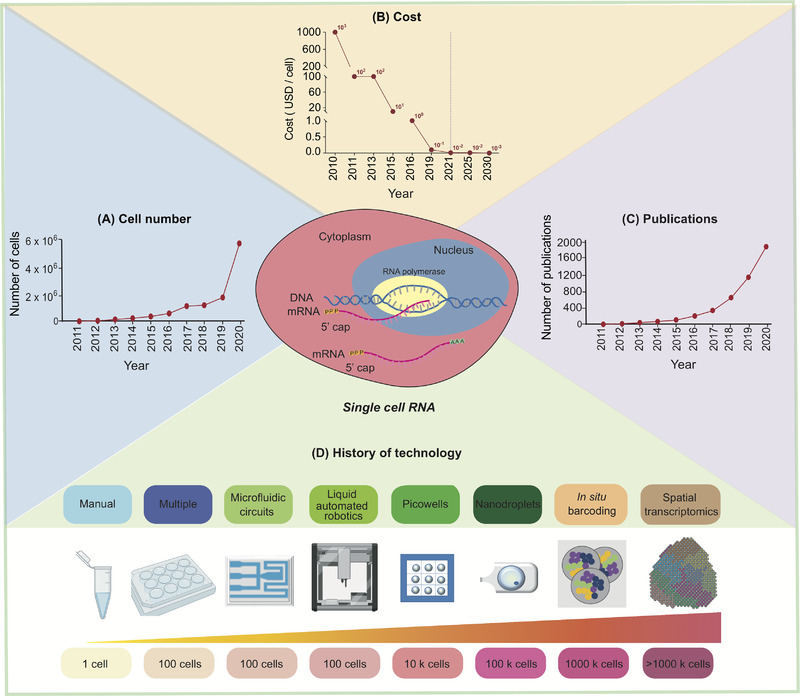
Figure 1. Development of scRNA-Seq [1]
The amount of RNA in a single mammalian cell is about 10 pg, of which only 1-5% is transcriptome RNA, far from the minimum standard for the construction of single-cell transcriptome libraries. To analyze gene expression activity in cells, the transcriptome requires a larger amount of starting RNA. Moreover, traditional transcriptome sequencing technologies introduce amplification biases and nucleic acid information loss during the process of mRNA reverse transcription into cDNA. Therefore, applying transcriptome analysis to single-cell analysis needs to address two main issues: acquiring single-cell and amplifying single-cell cDNA for library construction. The procedures of scRNA-seq mainly include single-cell isolation and capture, cell lysis, reverse transcription (conversion of their RNA into cDNA), cDNA amplification and library preparation.
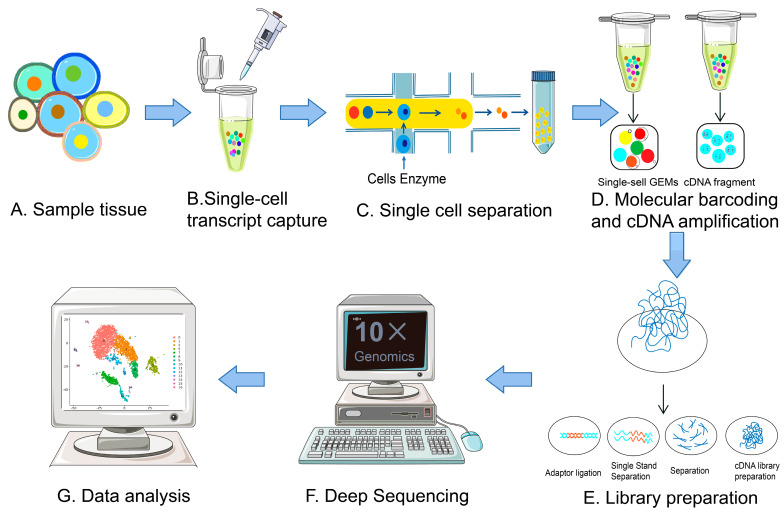
Figure 2. Single-Cell scRNA-Seq Experimental Workflow [2]
1. Applications of scRNA-Seq in Drug Discovery and Development
Single-cell technologies, particularly scRNA-seq, along with related computational tools and the growing available public data resources, are transforming drug discovery and development. Because of enhanced understanding of diseases on cellular subtypes, new opportunities for target identification arise, and high-throughput genomic screening combined with scRNA-seq is enhancing target identification and prioritization. scRNA-seq also aids in selecting relevant preclinical disease models and provides new insights into the mechanisms of drug action. In clinical development, scRNA-seq can inform decision-making via improved biomarker identification for patient stratification and more precise monitoring of drug responses and disease progression.
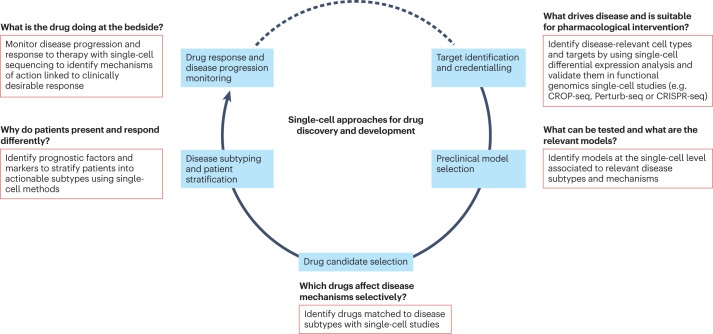
Figure 3. Applications of scRNA-Seq in Drug Development Process [3]
(1) Drug Screening and Drug Mode of Action (MoA) Analysis
High-throughput screening (HTS) in drug discovery is traditionally performed using coarse (cell viability or proliferation) or highly specific (marker expression) readouts. If an unbiased phenotypic assessment is chosen, bulk assessment (such as RNA-seq) assumes all cells behave similarly. In contrast, scRNA-seq provides a more detailed view of responding cell types and corresponding cell type-specific changes (pathways, off-target effects, dose-response profiles), thus allowing the separation of confounding factors such as the cell cycle. Therefore, HTS methods have recently been combined with scRNA-seq readouts. Standard HTS can test a large number of compounds but usually at a single dose and under very limited biological conditions, whereas new HTS methods using SC gene expression readouts can test multiple doses and conditions simultaneously and are highly suitable for drug MoA studies.
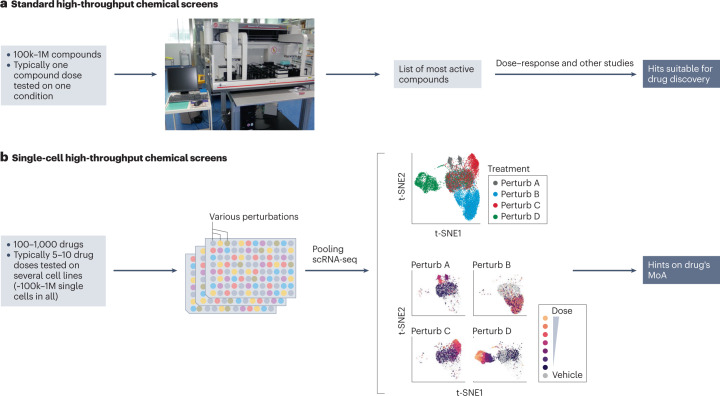
Figure 4. Single-Cell HTS [3]
(2) Biomarkers and Patient Stratification
In some cases, patients can be stratified into refined populations othe basis of disease prognosis or therapeutically relevant markers that predict drug response. These prognostic or predictive biomarkers are often used as eligibility criteria in clinical trials to identify patients more likely to exhibit disease progression or respond to a drug. scRNA-seq or single-cell multi-omics techniques can identify predictive biomarkers from cohorts of patients participating in earlphase clinical studies. This type of predictive biomarker can be used to identify patients who could benefit from a given treatment as a biomarker enrichment strategy.
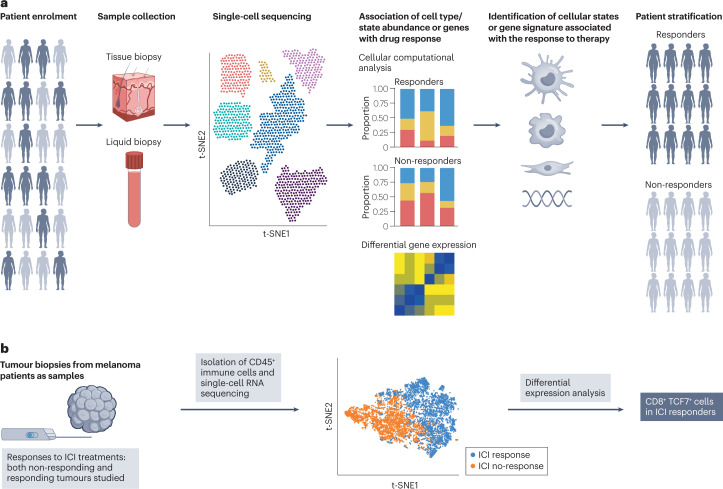
Figure 5. Application of scRNA-Seq in Biomarker Discovery and Patient Stratification [2]
(3) Monitoring Drug Response and Disease Progression
Using single-cell sequencing methods for clinical monitoring of disease progression and treatment responses has begun to impact clinical decision-making. Oncology is a leading field in this area. The concept of minimal residual disease (MRD) is a metric for measuring remaining cancer cells during or after treatment and has always been central to assessing drug response. For example, patients with acute myeloid leukemia (AML) often carry multiple subclones, each with complex molecular abnormalities. Current clinical practice defines complete remission as <5% blasts detected through bone marrow morphological assessment, without evaluating subclone molecular abnormalities or their evolution during treatment. Increasing evidence suggests that MRD assessment results below the 5% threshold are a risk factor for relapse, thus providing guidance for treatment decisions. Single-cell sequencing mutational mapping for MRD assessment (unlike more traditional MRD methods) can evaluate subclones at lower detection limits and analyze the evolution of subclones throughout the treatment process. Single-cell sequencing mutation profiling enhances the sensitivity and specificity of MRD detection and can also identify drug-resistant clones leading to relapse.
The associated risk of relapse partially explained by the persistence of resister cells. This type of drug resistance is typically driven by non-genetic adaptive mechanisms, although little is known about these mechanisms. To study rare and transient drug-resistant persister cells, a high-complexity lentiviral barcode library named Watermelon was developed to simultaneously track the clonal lineage, proliferation status, and transcriptome of individual cells during drug treatment. This method identified rare cancerous persistent lineages that are preferentially poised to proliferate under drug pressure and found that upregulation of antioxidant gene programmes and a metabolic shift to fatty acid oxidation were associated with persister proliferative capacity.
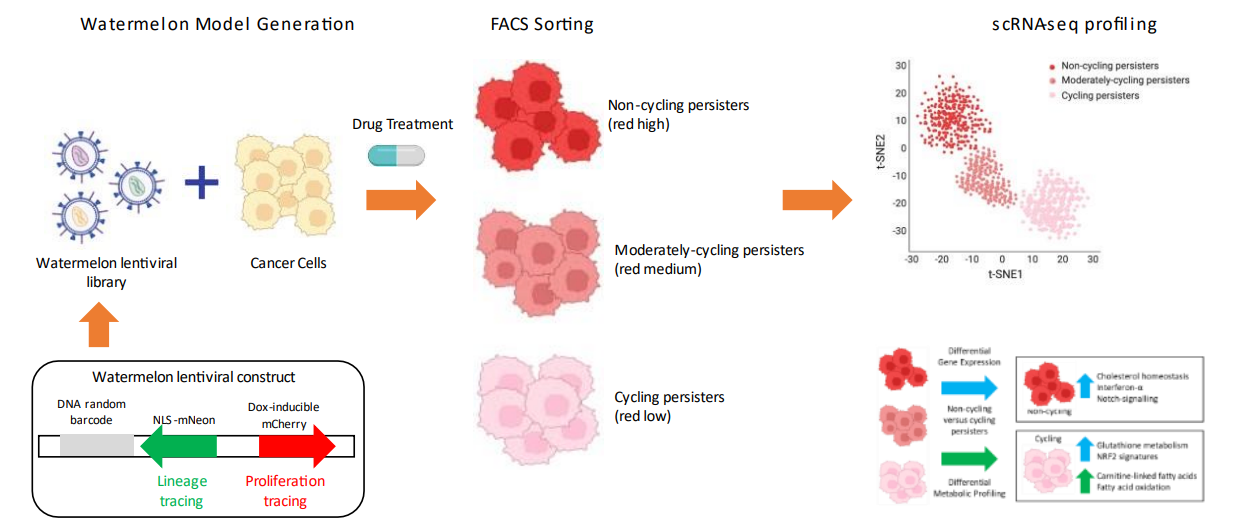
Figure 6. Watermelon Method Principle [2]
2. Unveiling Heterogeneity in Tissue and Disease
scRNA-Seq serves as a pivotal technique for elucidating cellular metabolites and gene expression, primarily utilized to explore the dynamics between cell subpopulations and gene regulatory mechanisms. This methodology finds application across various fields including embryology, tissue and organ development, oncology, and immunology.
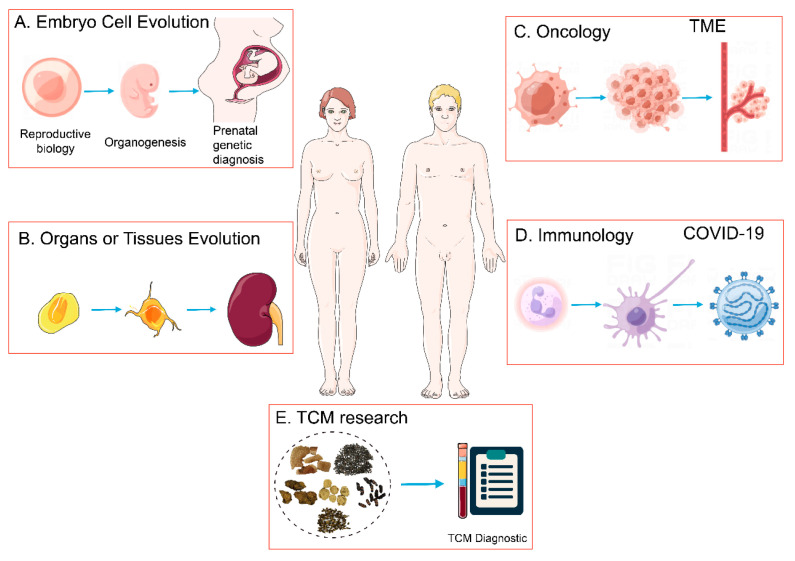
Figure 7. Applications of scRNA-Seq [1]
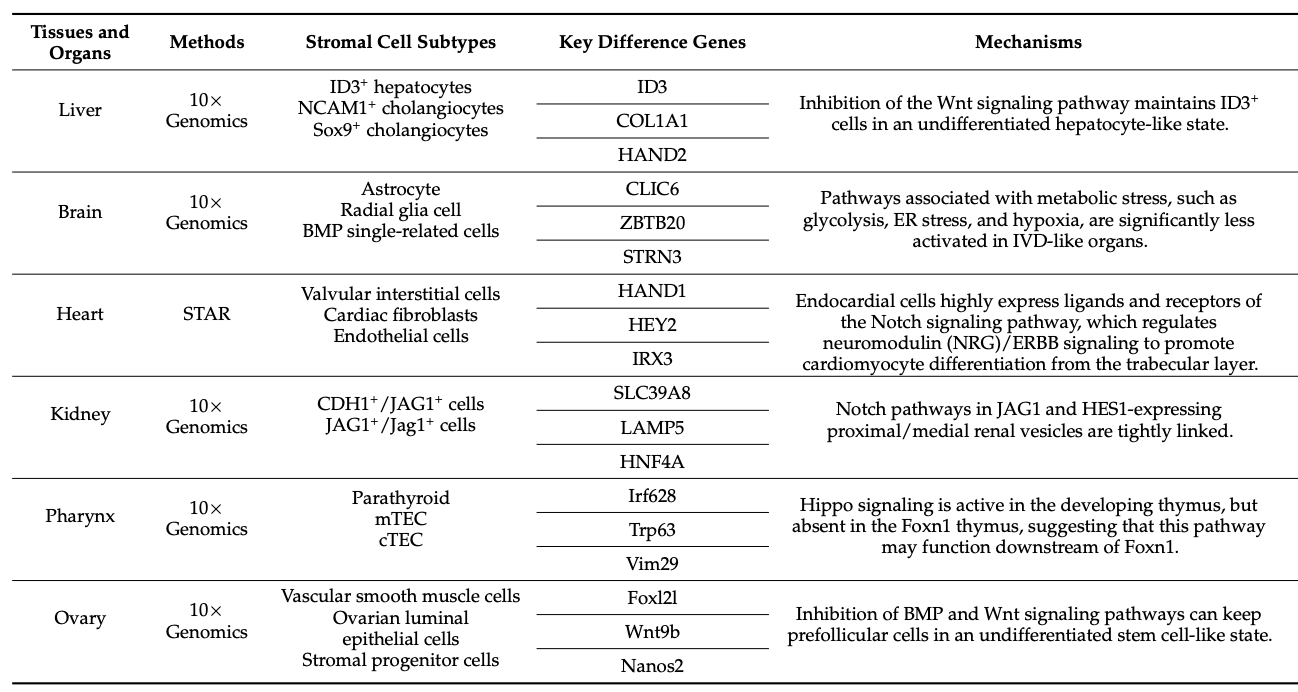
Table 1. Advances in Tissue Research Using scRNA-Seq [1]
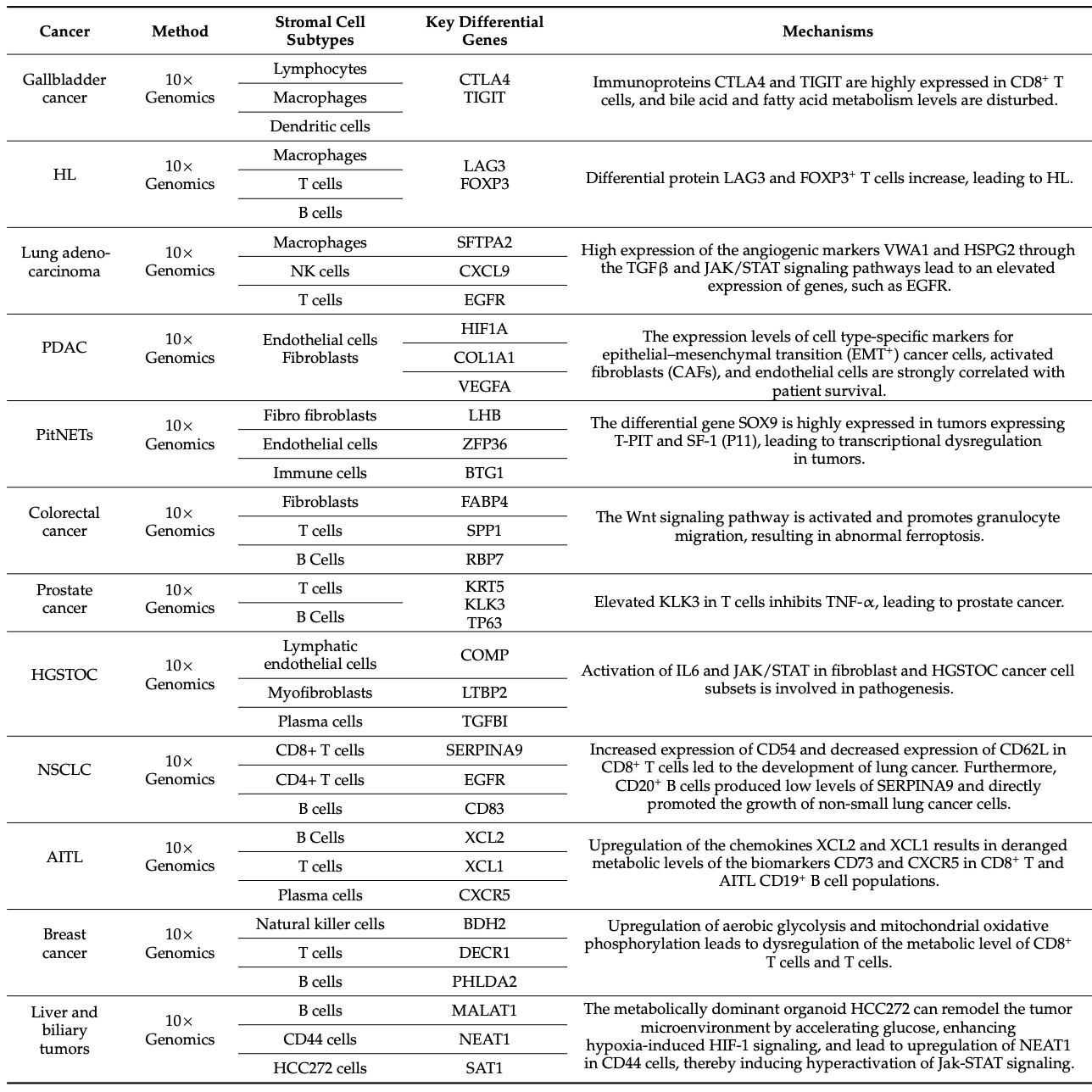
Table 2. Advances in Cancer Research Using scRNA-Seq [1]
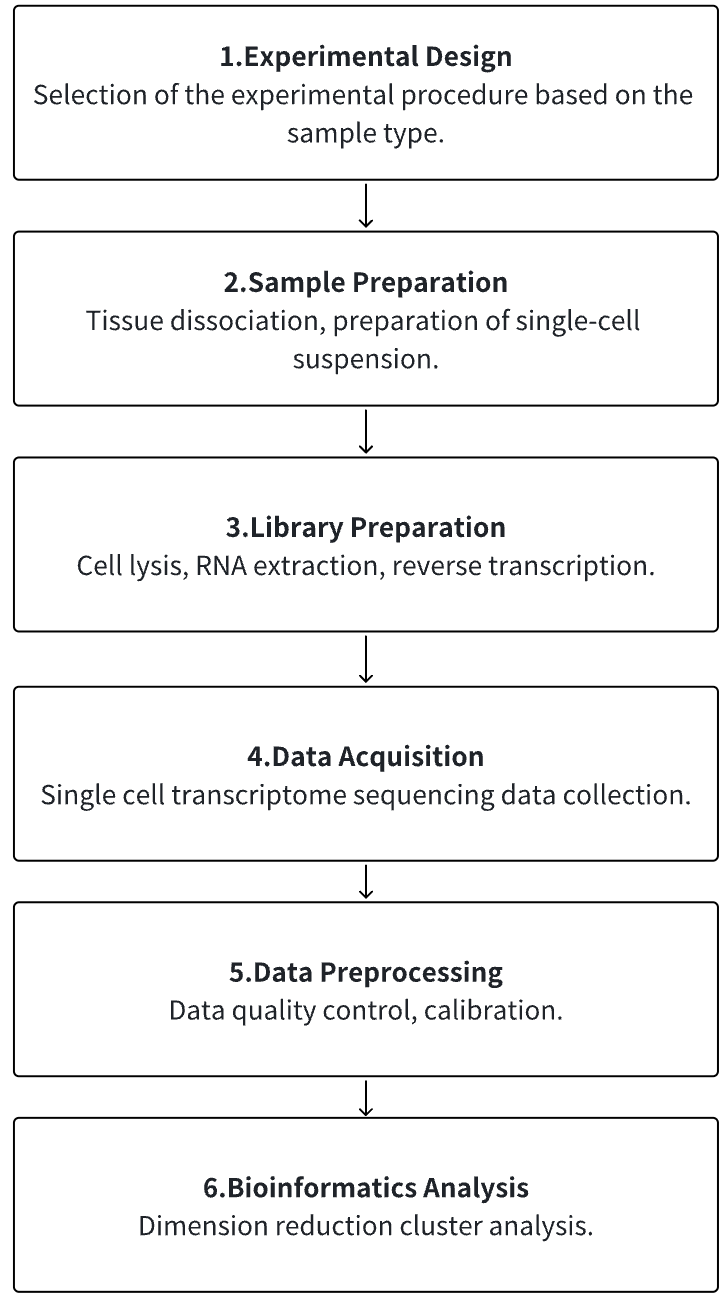
Analysis Workflow
1. According to the Experimental Requirements to Determine the Experimental Process
2. Tissue Dissociation, Preparation of Single-cell Suspension
3. Cell Lysis, RNA Extraction, Reverse Transcription
4. Single Cell Transcriptome Sequencing Data Collection
5. Data Preprocessing
6. Advanced Bioinformatics Analysis
Service Advantages
1. Adaptation to Multiple Types of Sample Detection
2. High-Reliability, High-Precision Single Cell Sequencing Service
3. Comprehensive Bioinformatics Analysis
Sample Results
1. Integration of single-cell transcriptome analysis has unveiled new insights into post-COVID-19 pulmonary fibrosis and potential therapeutic targets.
The ongoing global pandemic has resulted in numerous patients suffering from persistent symptoms such as post-COVID pulmonary fibrosis (PCPF). Research leveraged scRNA-Seq data from lung tissues of COVID-19, idiopathic pulmonary fibrosis (IPF) patients, and a rat transforming growth factor beta‐1‐induced fibrosis model treated with antifibrotic drugs to identify novel targets for PCPF treatment. Alveolar macrophage counts in COVID-19 patients were lower compared to healthy controls, whereas monocyte-derived macrophage counts were elevated in both COVID-19 and IPF patients. Comparative transcriptomic analyses indicated a vital role for macrophages in the development and progression of both IPF and COVID-19, with an upregulation of fibrosis and inflammation-related genes. Enrichment analyses revealed increased inflammation and proteolysis activities, with a decrease in ribosome biogenesis. Both macrophage types showed enhanced cholesterol efflux and glycolysis. These studies suggested that antifibrotic drugs could counteract critical mediators of pulmonary fibrosis in COVID-19, elucidated potential molecular mechanisms underlying severe cases and highlighted the therapeutic potential of these drugs in treating COVID-19. These insights were crucial for devising new strategies to treat PCPF.
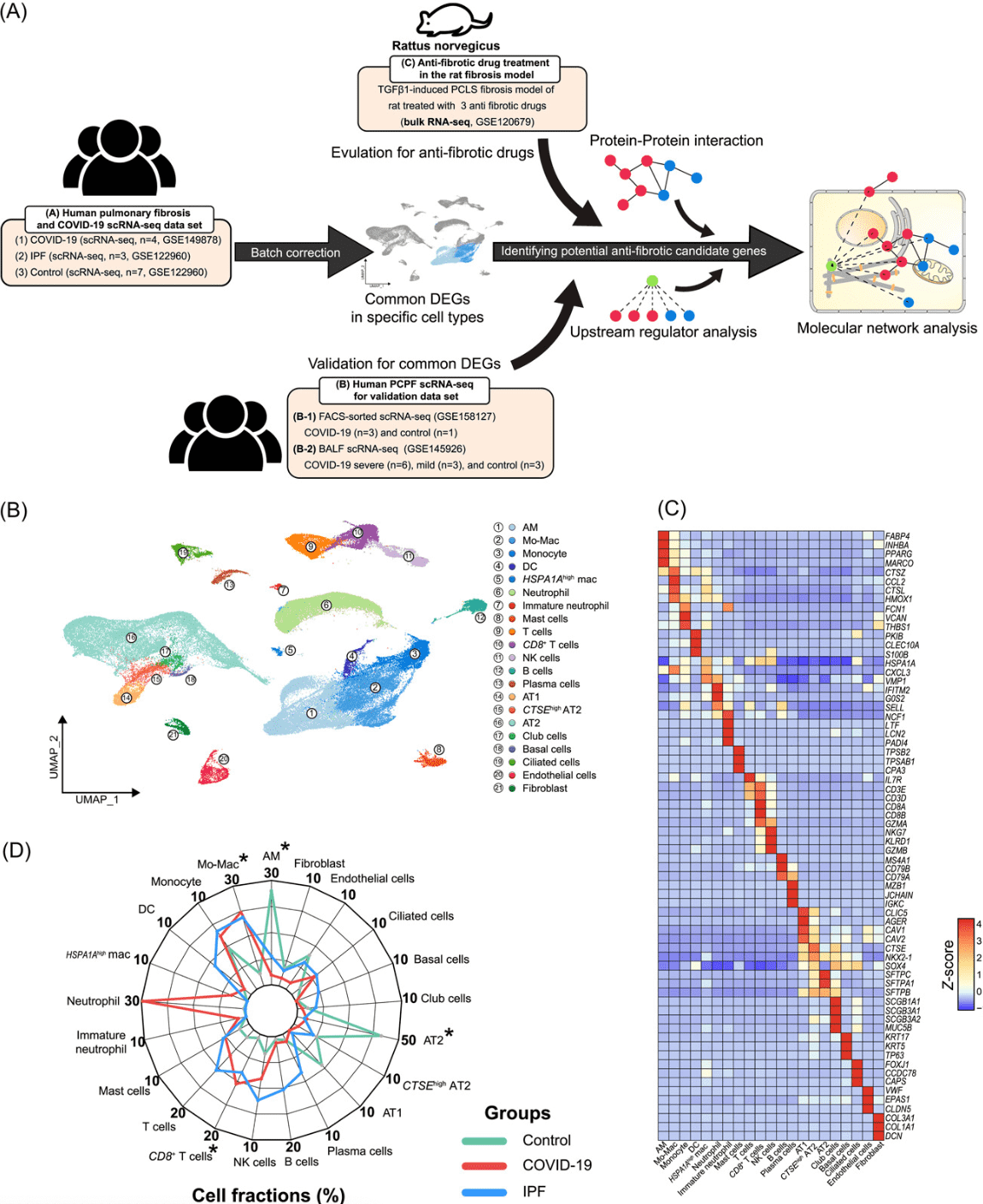
Figure 8. Similar Cellular Abundance Changes in Lung Tissues of COVID-19 and IPF Patients Suggest Potential Regulatory Cell Types in Fibrosis [5]
2. A single-cell analysis of breast cancer cell lines has been conducted to examine tumor heterogeneity and drug responses.
Cancer cells within the same tumor exhibit heterogeneous phenotypes and dynamic plasticity. The evaluation of this heterogeneity and its impact on outcomes and drug responses remains unclear. An analysis involved transcriptomic profiling of 35,276 individuals cells from 32 different breast cancer cell lines to creat a single-cell atlas. This research has highlighted the significant heterogeneity in biomarker expression. A deconvolution algorithm was developed and applied to this atlas to deduce cell line compositions from bulk gene expression data of tumor biopsies, facilitating patient stratification based on cell lineage. Moreover, linking large-scale in vitro drug screening outcomes with single-cell data has enabled the prediction of drug responses, starting from the single-cell atlas. This analysis demonstrated that transcriptional heterogeneity allowed cells with varying drug sensitivities to coexist within the same population, offering a framework for assessing tumor heterogeneity in terms of cell lineage composition and drug response.
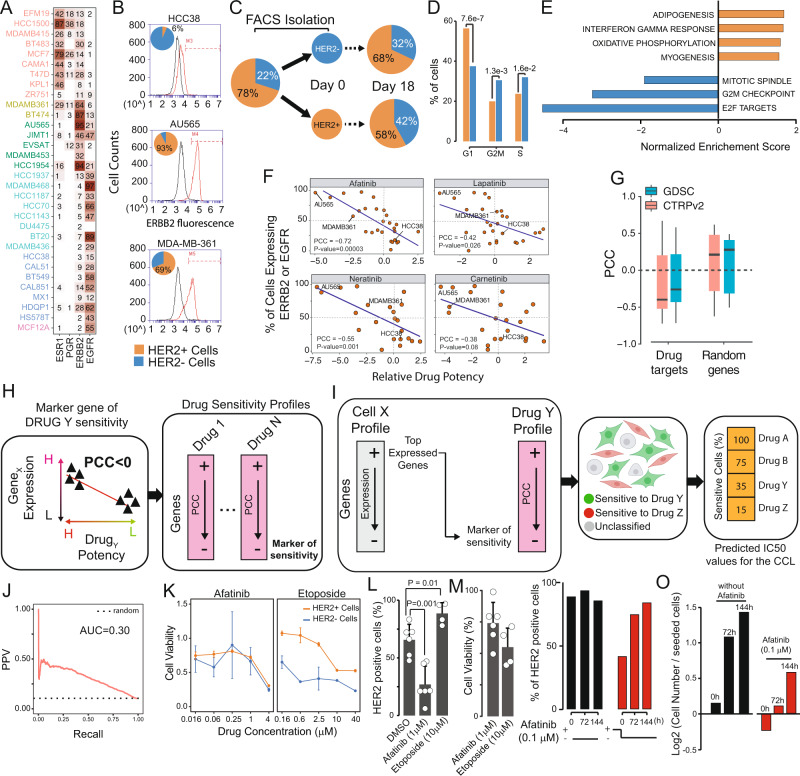
Figure 9. Impact of Transcriptional Heterogeneity on Drug Responses in Breast Cancer Cell Lines [6]
3. scRNA-Seq has been employed to deconvolve drug responses specific to different cell types within human tumor tissues.
Preclinical studies require models that recapitulate the cellular diversity of human tumors and offer insights into the drug sensitivities of specific cellular populations. Ideally, a platform enabled rapid screening of cell type-specific drug sensitivities directly within patient tumor tissues and unveiled strategies to overcome intratumoral heterogeneity. This approach was applied in multiple drug perturbation studies on acute slices from six glioblastoma multiforme (GBM) resections to identify conserved drug responses, and in an additional GBM surgery to determine patient-specific responses. Acute slice cultures were demonstrated to effectively recapitulate the cellular and molecular characteristics of the original tumor tissues and affirmed the feasibility of drug screening directly from individual tumors. Detailed investigations of etoposide and the histone deacetylase (HDAC) inhibitor Pabinostat in these cultures highlighted cell-type specific responses across several patients. Etoposide consistently affected proliferating tumor cells, while Pabinostat treatment influenced both tumor and non-tumor populations, including unexpected impacts on the immune microenvironment.
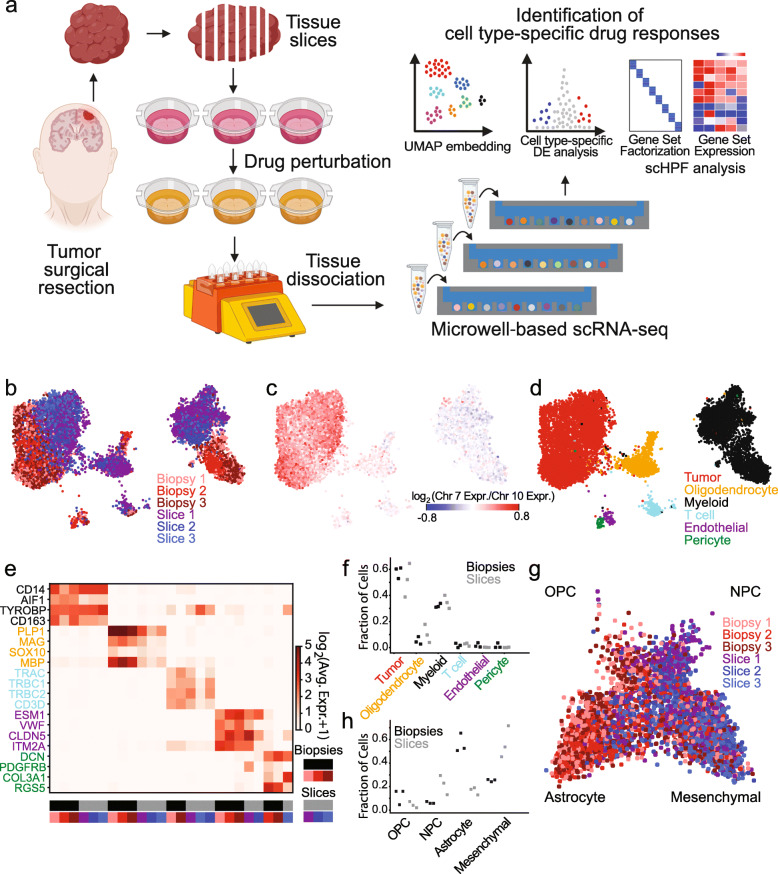
Figure 10. Experimental and Analytical Methods for Drug scRNA-Seq in Tissue Slice Cultures [7]
Sample Submission Requirements
1. Cell Quantity and Concentration: It is recommended to have 5 × 105 cells and a concentration of 1000 cells/ μ L.
2. Cell Activity: Greater than 80%, it is recommended to build a risk library of 105.
3. Cell Diameter: 5 μ M ≤ X ≤ 30 μ M.
4. Efforts should be made to minimize impurity contamination.
Services at MtoZ Biolabs
1. The Complete Experimental Procedure
2. Relevant Instrument Parameters
3. Original Experimental Data
4. Data Analysis Reports
References
[1] Jovic D, Liang X, Zeng H, Lin L, Xu F, Luo Y. Single-cell RNA sequencing technologies and applications: A brief overview. Clin Transl Med. 2022 Mar;12(3):e694. doi: 10.1002/ctm2.694. PMID: 35352511; PMCID: PMC8964935.
[2] Wang S, Sun ST, Zhang XY, Ding HR, Yuan Y, He JJ, Wang MS, Yang B, Li YB. The Evolution of Single-Cell RNA Sequencing Technology and Application: Progress and Perspectives. Int J Mol Sci. 2023 Feb 2;24(3):2943. doi: 10.3390/ijms24032943. PMID: 36769267; PMCID: PMC9918030.
[3] Van de Sande B, Lee JS, Mutasa-Gottgens E, Naughton B, Bacon W, Manning J, Wang Y, Pollard J, Mendez M, Hill J, Kumar N, Cao X, Chen X, Khaladkar M, Wen J, Leach A, Ferran E. Applications of single-cell RNA sequencing in drug discovery and development. Nat Rev Drug Discov. 2023 Jun;22(6):496-520. doi: 10.1038/s41573-023-00688-4. Epub 2023 Apr 28. PMID: 37117846; PMCID: PMC10141847.
[4] Kui L, Kong Q, Yang X, Pan Y, Xu Z, Wang S, Chen J, Wei K, Zhou X, Yang X, Wu T, Mastan A, Liu Y, Miao J. High-Throughput In Vitro Gene Expression Profile to Screen of Natural Herbals for Breast Cancer Treatment. Front Oncol. 2021 Aug 13;11:684351. doi: 10.3389/fonc.2021.684351. PMID: 34490085; PMCID: PMC8418118.
[5] Kim Y, Kim Y, Lim HJ, Kim DK, Park JH, Oh CM. Integrative single-cell transcriptome analysis provides new insights into post-COVID-19 pulmonary fibrosis and potential therapeutic targets. J Med Virol. 2023 Nov;95(11):e29201. doi: 10.1002/jmv.29201. PMID: 37966390.
[6] Gambardella G, Viscido G, Tumaini B, Isacchi A, Bosotti R, di Bernardo D. A single-cell analysis of breast cancer cell lines to study tumour heterogeneity and drug response. Nat Commun. 2022 Mar 31;13(1):1714. doi: 10.1038/s41467-022-29358-6. PMID: 35361816; PMCID: PMC8971486.
[7] Zhao W, Dovas A, Spinazzi EF, Levitin HM, Banu MA, Upadhyayula P, Sudhakar T, Marie T, Otten ML, Sisti MB, Bruce JN, Canoll P, Sims PA. Deconvolution of cell type-specific drug responses in human tumor tissue with single-cell RNA-seq. Genome Med. 2021 May 11;13(1):82. doi: 10.1186/s13073-021-00894-y. PMID: 33975634; PMCID: PMC8114529.
How to order?

