





HDX Based Protein Conformation Analysis Service
Hydrogen-deuterium exchange mass spectrometry (HDX-MS) is a mass spectrometry (MS) technique that studies protein spatial conformation. In this process, protein hydrogen atoms can exchange with deuterium atoms in heavy water. The hydrogen atoms of amide bonds on the protein surface or those involved in protein-ligand interactions exchange at a faster rate than those deeper within the protein or not involved in interactions. By measuring the rate of hydrogen-deuterium exchange across different protein segments through MS, information about changes in protein conformation can be deduced. HDX-MS is valuable for analyzing the folding, oligomerization, and conformational changes of proteins in solution; it is also used to detect protein interactions with other molecules, including protein-protein, protein-peptide, protein-small molecule, protein-nucleic acid, and protein-membrane interactions.
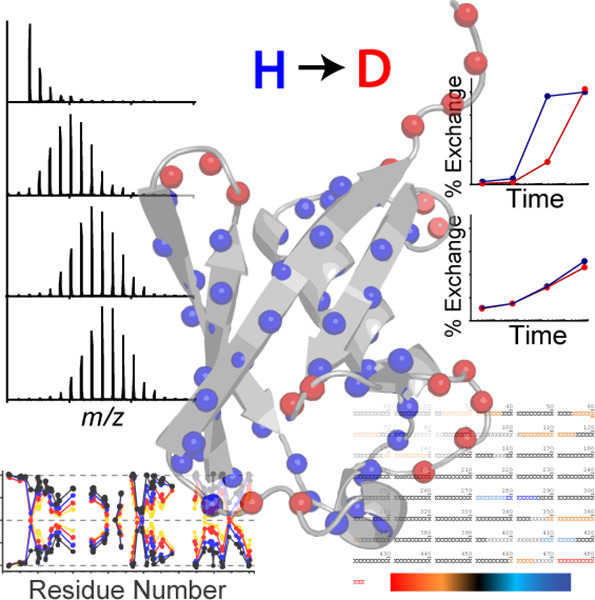
Figure 1. Protein HDX-MS [1]
The typical methodology in most HDX-MS studies involves transferring the protein from an aqueous buffer to a deuterium-enriched buffer under native conditions, initiating the exchange process. Due to the mass difference of approximately 1 Da between hydrogen (1H) and deuterium (2H), the incorporation of deuterium is easily detectable by MS. The fundamental principles of amide HDX, initially proposed by Linderstrom-Lang, still apply. Specific amides in the protein can be in either a protected or exposed state, influencing their ability to deprotonate. When local conformational fluctuations or global unfolding render the amide accessible, the hydrogen can undergo exchange with solvent at a specific rate, termed the chemical exchange rate (kch). Once exchanged, the protein may then revert back to a closed state. Hydrogen bonding is generally considered a primary determinant of amide protection, but that solvent occlusion also plays a role in determining protection from exchange.
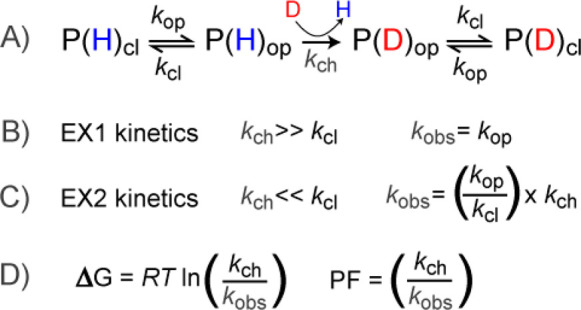
Figure 2. Basic Principles of Amide Exchange in Proteins [1]
Experimental approaches in HDX-MS are categorized into top-down and bottom-up strategies. All HDX-MS methodologies (excluding gas-phase HDX-MS) begin with a labeling step in aqueous conditions, where the protein is exposed to deuterium for a predetermined time before quenching. The protein is then injected into a mass spectrometer for full mass measurement; in the instrument, the intact protein is partially fragmented to obtain full structural domains or individual peptide segments for comparative analysis in different states (top-down). Alternatively, proteins are digested with proteases in a fluid system to produce peptides for mass spectrometric analysis (bottom-up). This bottom-up method can be further refined either by calculating individual exchange rates using mathematical approaches (pseudo amino-acid resolution) or by targeted electron capture dissociation (ETD) fragmentation of individual peptides (middle-down).
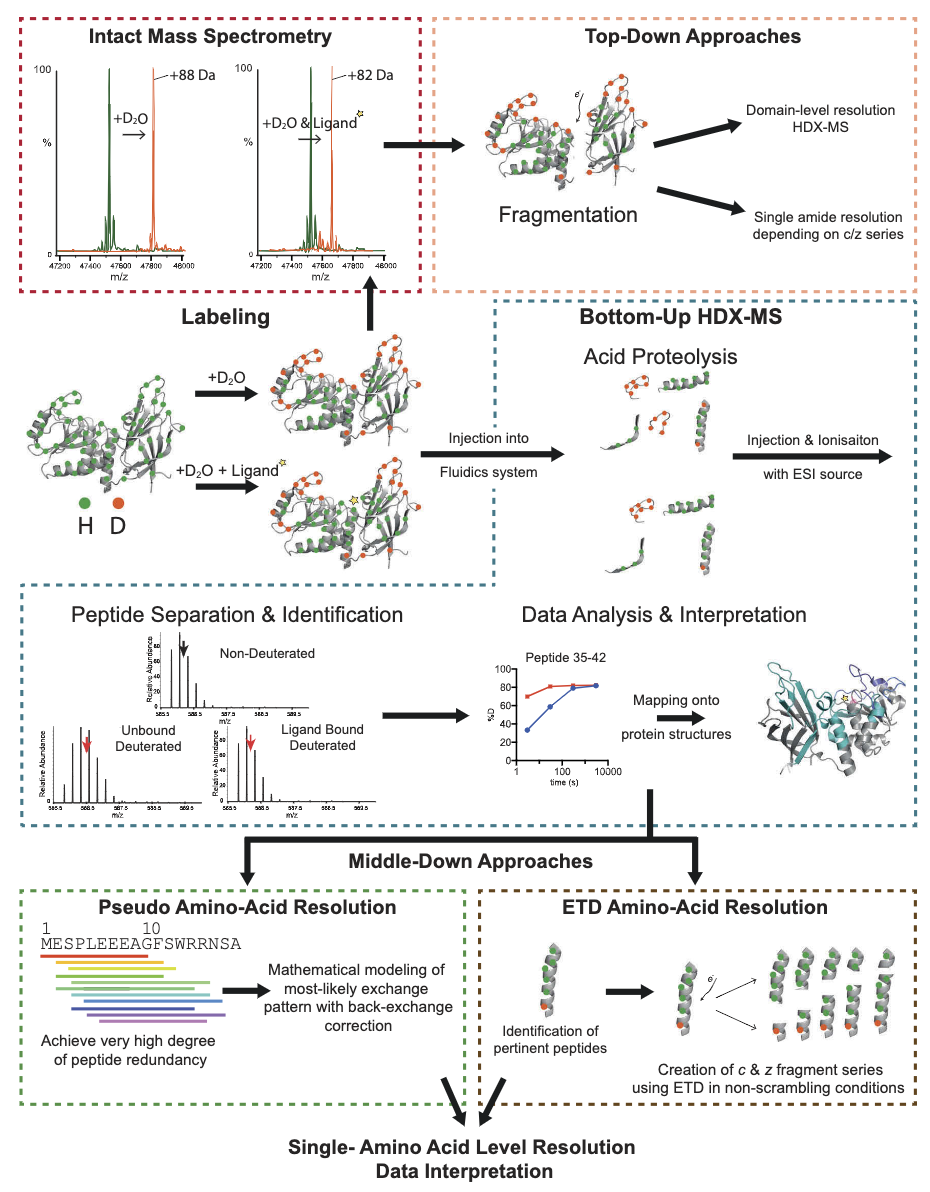
Figure 3. Experimental Strategies in HDX-MS [2]
One of the reasons HDX-MS has been so successful is its inherent versatility; it can be applied to almost any protein system, many of which are too large, flexible, heterogeneous, or limited in sample size for other structural tools. HDX-MS addresses key questions about the dynamics, conformation, and interactions of biotherapeutic proteins, including epitope mapping, protein-drug binding, protein-protein interactions, aggregation, effects of mutations on protein conformation and dynamics, and configurational changes due to formulation and stability testing.
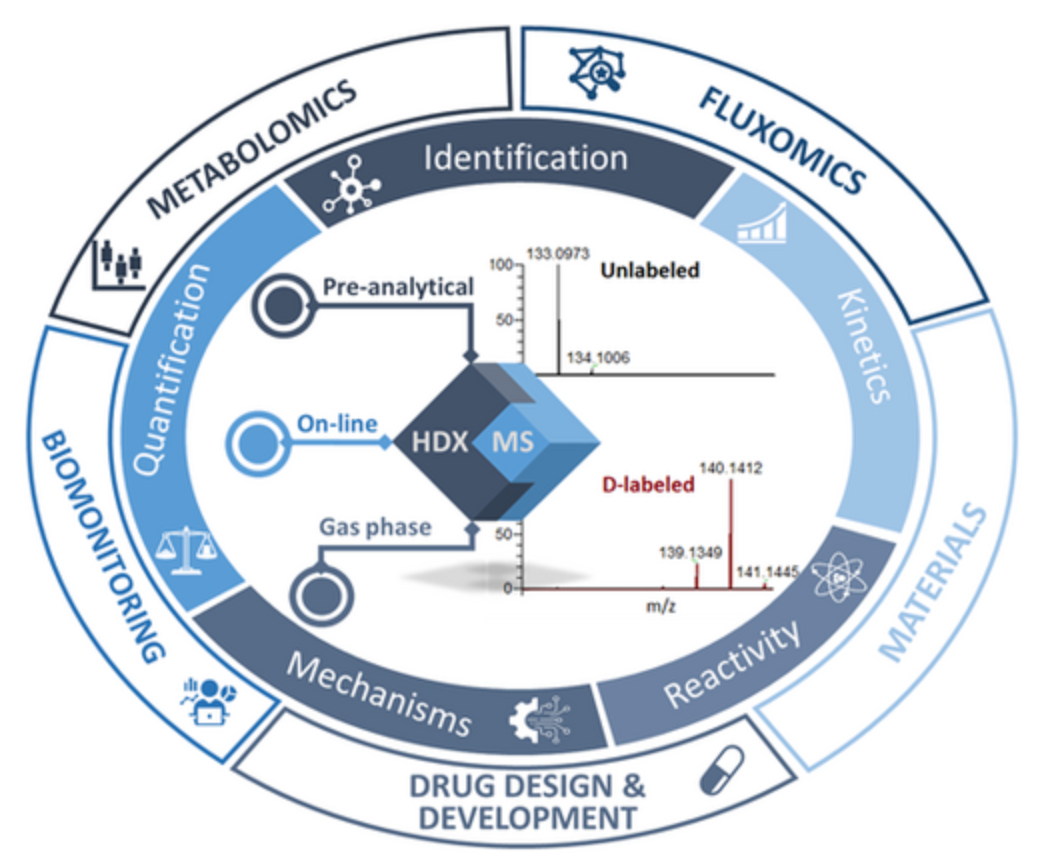
Figure 4. Applications of HDX-MS [3]
MtoZ Biolabs offers a comprehensive HDX-MS platform, providing services such as protein-antibody interaction analysis, protein-small molecule interaction identification, and protein-protein interaction site mapping. Our team can express and purify substrate proteins using eukaryotic/prokaryotic expression systems, perform hydrogen-deuterium exchange labeling post-binding of substrate proteins with small molecules, and conduct high-resolution mass spectrometric analysis using either a top-down or bottom-up approach to identify conformational changes and interaction epitopes. MtoZ Biolabs provides a one-stop solution for protein conformation and epitope characterization, including protein interaction experiments, HDX-MS sample preparation, mass spectrometry analysis, and data analysis services.
References
[1] James EI, Murphree TA, Vorauer C, Engen JR, Guttman M. Advances in Hydrogen/Deuterium Exchange Mass Spectrometry and the Pursuit of Challenging Biological Systems. Chem Rev. 2022 Apr 27;122(8):7562-7623. doi: 10.1021/acs.chemrev.1c00279. Epub 2021 Sep 7. PMID: 34493042; PMCID: PMC9053315.
[2] Masson GR, Jenkins ML, Burke JE. An overview of hydrogen deuterium exchange mass spectrometry (HDX-MS) in drug discovery. Expert Opin Drug Discov. 2017 Oct;12(10):981-994. doi: 10.1080/17460441.2017.1363734. Epub 2017 Aug 17. PMID: 28770632.
[3] Damont A, Legrand A, Cao C, Fenaille F, Tabet JC. Hydrogen/deuterium exchange mass spectrometry in the world of small molecules. Mass Spectrom Rev. 2023 Jul-Aug;42(4):1300-1331. doi: 10.1002/mas.21765. Epub 2021 Dec 2. PMID: 34859466.
How to order?

