





Comparative Proteomics Service
Comparative proteomics is a powerful approach that systematically analyzes the differences in protein composition, expression levels, and post-translational modifications between biological samples using high-throughput techniques such as mass spectrometry. It provides an unbiased and comprehensive means to reveal dynamic changes in proteins under varying conditions, helping to understand the roles of proteins in complex biological processes and their connections to the onset and progression of diseases. The purpose of comparative proteomics is to uncover the molecular mechanisms underlying biological systems under different environmental stimuli, disease states, or genetic backgrounds by comparing proteomes across sample groups. It can identify key regulatory proteins, elucidate changes in signaling pathways, and explore novel biomarkers and drug targets, offering valuable insights for both basic research and clinical translation. Comparative proteomics addresses critical questions such as identifying disease-associated proteins, analyzing functional changes in metabolic pathways, and uncovering molecular responses to external stimuli. Through precise protein quantification and functional analysis, comparative proteomics provides unprecedented resolution and depth for understanding complex biological phenomena, driving advancements in life science research and personalized medicine.
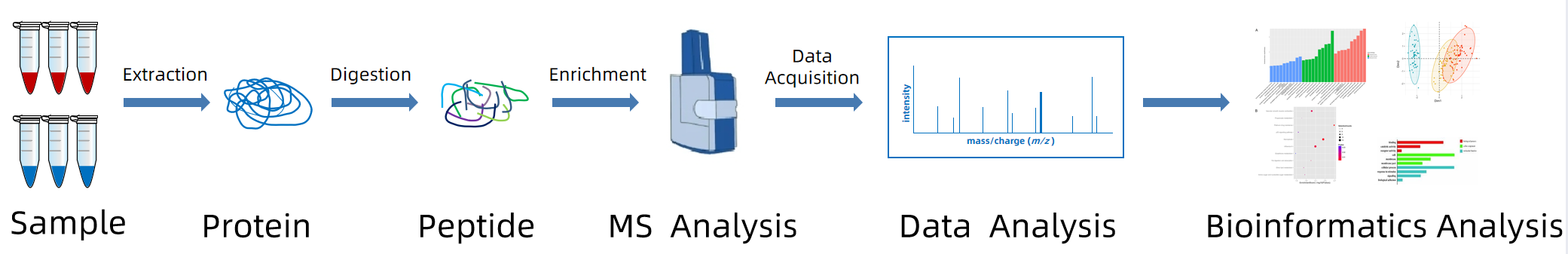
Figure 1. The Workflow of Comparative Proteomics
MtoZ Biolabs, an integrated Chromatography and Mass Spectrometry (MS) Services Provider, provides advanced proteomics, metabolomics, and biopharmaceutical analysis services to researchers in biochemistry, biotechnology, and biopharmaceutical fields. Our ultimate aim is to provide more rapid, high-throughput, and cost-effective analysis, with exceptional data quality and minimal sample consumption. Utilizing advanced mass spectrometry platforms, MtoZ Biolabs offers high-precision comparative proteomics services to comprehensively analyze protein expression differences and functional changes across various sample conditions. Our professional team, backed by extensive project experience, provides end-to-end support, from sample preparation and protein quantification to data analysis, ensuring high-quality and highly reproducible results to empower researchers in their studies. If you are interested in our service, please contact us freely.
Case Study
1. Comparative Proteomics of Saliva of Healthy and Gingivitis Individuals from Rio de Janeiro
In this study, researchers identified human and bacterial proteomes from the saliva of individuals with gingivitis or healthy volunteers. The reported cohort included 18 participants (6 with gingivitis and 12 healthy controls). Comparative proteomics was performed using quantitative mass spectrometry. A total of 74 human proteins and 116 bacterial proteins were identified in saliva. The primary functional categories altered in the human proteome were immune response, followed by transport and protease inhibition. In the bacterial proteome, most identified proteins originated from Fusobacteria, followed by Chlamydiae and Spirochaetes. Statistically significant differences were observed between the groups. The 15 most important human proteins influencing the differences between the case and control groups included cystatin S, α-amylase, lactotransferrin, and elongation factor E. Researchers suggested that bacterial proteins from Porphyromonas gingivalis and Fusobacterium nucleatum subspecies, associated with the red and orange complexes, are closely linked to the development of periodontal disease.
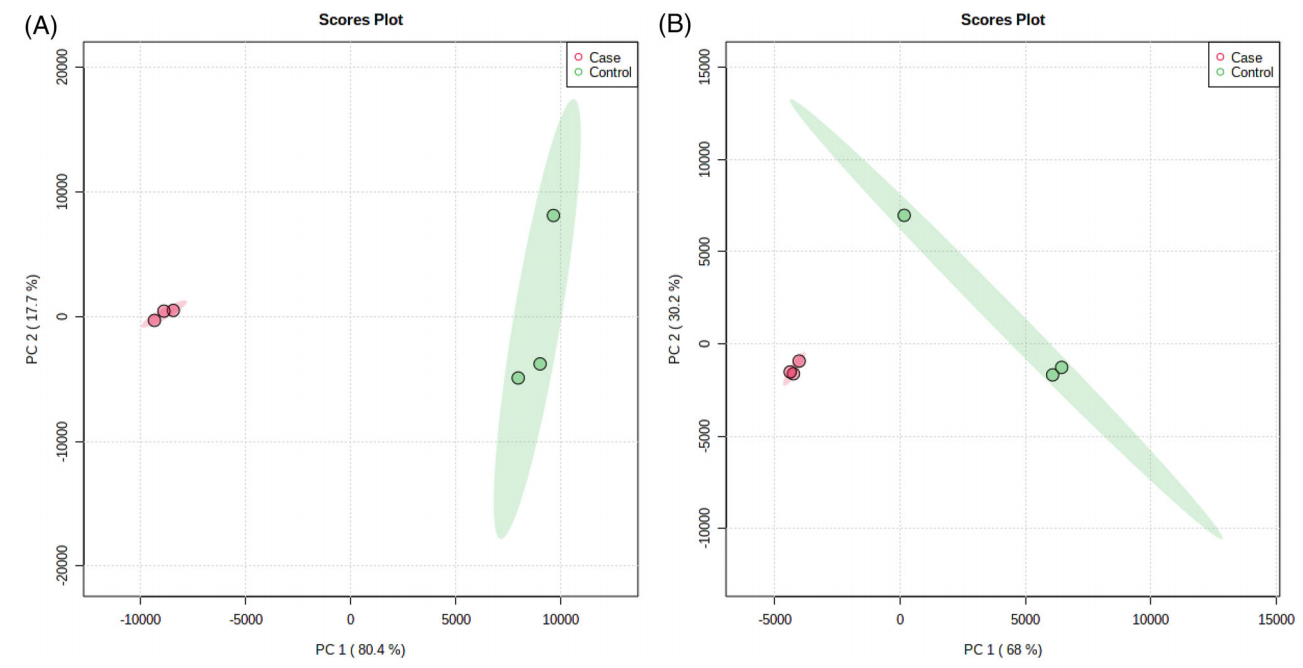
Da, Silva, CVF. et al. Proteomics Clin Appl. 2023.
Figure 2. Principal Component Analysis of Case and Control Groups of Human and Bacterial Proteins
2. Comparative Proteomics Analysis for Elucidating the Interaction Between Host Cells and Toxoplasma gondii
Toxoplasma gondii is a representative model organism of the phylum Apicomplexa that can infect nearly all warm-blooded animals, including humans. Host-parasite interactions are critical for T. gondii to invade host cells and complete its life cycle. In this study, researchers conducted comparative proteomics based TMT to investigate global proteomic changes during intracellular infection in host cells [human foreskin fibroblasts (HFF): infected HFF (HT) vs. uninfected HFF (H)] and in T. gondii itself [HT vs. T. gondii (T)]. Overall, 3,477 and 1,434 proteins were quantified, with 375 and 1,099 proteins exhibiting differential expression in host cells and T. gondii, respectively (adjusted p-value < 0.05 and fold change > 1.5 or < 0.67). The invasion of host cells by T. gondii relies on the secretion of numerous proteins from three specialized secretory organelles: micronemes, rhoptries, and dense granules. In the HT vs. T group, only a few secretory proteins were upregulated, such as microneme proteins (MIC: MIC6, MIC10), rhoptry proteins (ROP: ROP5, ROP17), and dense granule proteins (GRA: GRA4, GRA5, GRA12). In contrast, dozens of known secretory proteins were significantly downregulated in HFF infected by T. gondii. In HFF, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis revealed that differentially expressed proteins (DEPs) were enriched in metabolic processes and immune-related signaling pathways, such as NF-kB, cAMP, and Rap1 signaling pathways. Meanwhile, in T. gondii, DEPs were involved in ribosome biogenesis, the citric acid cycle, and galactose metabolism, suggesting altered cellular biosynthesis and metabolism following host cell invasion. These findings unveil novel modifications in the proteomes of host cells and T. gondii, enhancing our understanding of the underlying mechanisms of host-parasite interactions.
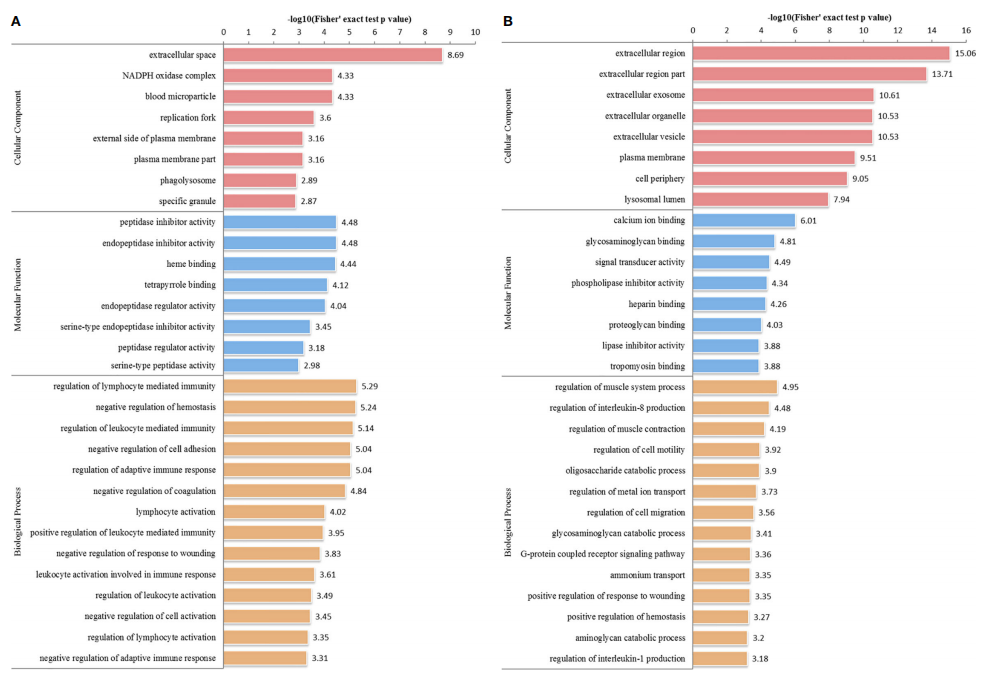
Sun, H. et al. Front Cell Infect Microbiol. 2021.
Figure 3. GO Distribution of Differentially Expressed Proteins in the HT vs. H Group
Service Advantages
Advantages of MtoZ Biolabs comparative proteomics service:
1. Comprehensive Detection of Low-Abundance Proteins
Leveraging advanced mass spectrometry platforms, including the Thermo Fisher Orbitrap series, MtoZ Biolabs excels in accurately identifying low-abundance proteins within complex samples, avoiding interference from high-abundance proteins and enabling deep insights into critical biological processes.
2. Accurate Quantification and High-Throughput Analysis
Utilizing multiple quantification strategies such as TMT, iTRAQ labeling, and label-free quantification, our comparative proteomics service ensures high sensitivity while delivering reliable and accurate protein quantification across experimental groups, meeting the needs of large-scale, high-throughput sample comparisons.
3. Expert Data Analysis and Biological Interpretation
With a dedicated bioinformatics team, MtoZ Biolabs employs cutting-edge analysis tools and databases to provide comprehensive support, from identifying differential proteins to functional enrichment analysis, uncovering biologically meaningful proteomic differences for clients.
4. Rigorous Standardization and High Reproducibility
Internationally recognized quality control standards and extensive project experience ensure consistent and reproducible experimental results, providing reliable data support for clients’ research.
FAQ
Q1: What strategies can be employed to improve the detection and quantification of low-abundance proteins in comparative proteomics, particularly when dealing with highly complex biological samples?
Answer: Improving the detection and quantification of low-abundance proteins in comparative proteomics requires a combination of optimized sample preparation, advanced instrumentation, and robust data analysis. Efficient sample fractionation techniques, such as high-resolution liquid chromatography, can reduce sample complexity and enhance protein detectability. Utilizing cutting-edge mass spectrometry platforms with high sensitivity and resolution, such as Orbitrap or FTICR-MS, ensures accurate identification of low-abundance proteins. Additionally, implementing enrichment methods for specific protein classes or modifications further increases detection rates. On the data analysis front, leveraging advanced bioinformatics tools and statistical approaches minimizes noise and false positives, enabling more precise quantification. Together, these strategies address the challenges posed by dynamic protein abundance ranges in complex biological samples.
Deliverables
1. Comprehensive Experimental Details
2. Materials, Instruments, and Methods
3. Relevant Liquid Chromatography and Mass Spectrometry Parameters
4. The Detailed Information of Proteomics
5. Mass Spectrometry Image
6. Raw Data
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

