





How to Obtain Comprehensive Protein Information Through de Novo Sequencing
The goal of de novo sequencing is to analyze the complete amino acid sequence of proteins from mass spectrometry data without bias, including information about mutations, post-translational modifications (PTMs), and isoforms. In order to obtain a comprehensive understanding of protein information, strategies such as optimizing sample preparation, using advanced mass spectrometry techniques, applying multiple fragmentation modes, utilizing intelligent data analysis, and performing PTM analysis must be combined to ensure the most complete analysis of protein sequences and their functional characteristics. This article systematically explores how to obtain comprehensive protein information through de novo protein sequencing.
Sample Preparation
Protein purity, integrity, and homogeneity are essential for obtaining comprehensive protein information. Sample preparation should prevent degradation, optimize proteolysis strategies, and remove non-protein contaminants.
1. Improving Protein Purity
(1) Removal of high-abundance proteins: High-abundance background proteins, such as albumin and IgG, should be removed using methods like immunoaffinity purification (IP), ultracentrifugation, or gel filtration, to highlight the signal of low-abundance target proteins.
(2) Removal of non-protein impurities: Impurities such as salts, detergents, and nucleic acids in the sample can interfere with mass spectrometry analysis. These can be effectively removed through techniques like dialysis or ultrafiltration.
2. Multi-Protease Digestion to Improve Sequence Coverage
(1) Combined protease digestion: Trypsin, when used alongside other proteases like Glu-C, Asp-N, and Lys-C, generates complementary peptide fragments that improve the coverage of regions that are difficult to sequence.
(2) Special protease selection: For structurally complex or difficult-to-degrade proteins, such as membrane proteins, nonspecific proteases (e.g., Proteinase K) combined with denaturants or auxiliary digestion methods can be employed.
Mass Spectrometry Analysis
1. Selection of an Appropriate High-Resolution Mass Spectrometer
(1) High-resolution Orbitrap or FT-ICR MS (Resolution > 100,000) provides high mass accuracy at the parts per million (ppm) level, which significantly enhances the ability to distinguish between amino acids.
(2) Q-TOF MS (Time-of-Flight Mass Spectrometry) combines high sensitivity with a wide dynamic range, making it suitable for analyzing complex protein samples.
2. Utilization of Multiple Fragmentation Modes
The optimization of fragmentation modes directly influences the integrity of the b and y ion series, which in turn impacts the accuracy of amino acid sequence derivation. The following fragmentation modes are commonly employed:
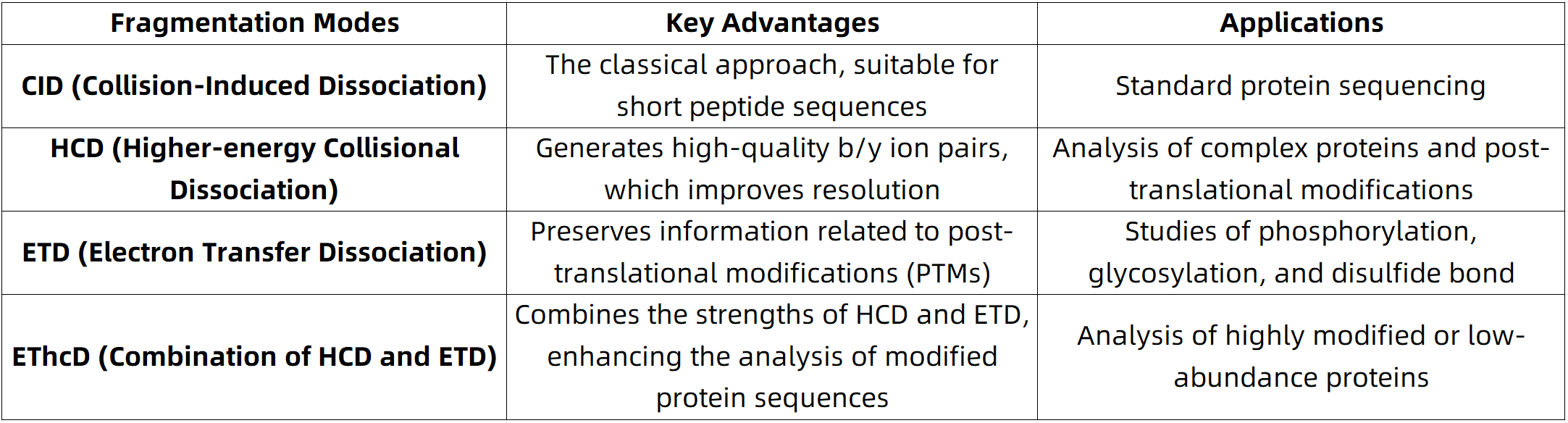
Figure 1
3. Intelligent Data Acquisition Strategy
(1) Data-dependent Acquisition (DDA): This method automatically selects the most abundant ion fragments for analysis, making it ideal for high-abundance proteins, where fragmentation patterns can be more easily detected and identified.
(2) Data-independent Acquisition (DIA): In contrast to DDA, DIA acquires data across the entire m/z range, allowing for more comprehensive analysis of low-abundance proteins, which might otherwise be missed in traditional data-dependent methods.
(3) Parallel Reaction Monitoring (PRM): This technique provides high-precision quantification of specific target proteins by monitoring selected ion fragments, thus significantly reducing background noise and improving specificity.
Data Analysis
1. AI-Driven Sequence Prediction
Traditional algorithms (such as PEAKS and pNovo+) rely on spectrum matching for sequence identification, but their accuracy is often limited by the quality of the mass spectrometry signals. In contrast, machine learning-based algorithms (such as DeepNovo and AlphaPept) leverage deep learning techniques to improve the assembly of longer peptide sequences and correct errors introduced during the sequencing process.
2. Differentiation and Correction of Amino Acid Isomers
(1) The differentiation of isomers such as Leucine (Leu) and Isoleucine (Ile) is assisted by high-resolution mass spectrometry and fragmentation techniques, such as electron transfer dissociation (ETD), which provide more detailed fragmentation patterns for these structurally similar amino acids.
(2) Chemical derivatization techniques, such as hydrogen/deuterium (H/D) exchange, further enhance the ability to distinguish between such isomers by providing additional structural information.
3. Data Integration and Spectrum Correction
(1) Combining peptide information obtained from digestions with different proteases enables a more comprehensive sequence assembly, compensating for any regions missed during digestion with a single protease.
(2) Advanced algorithms for spectrum quality enhancement and cross-validation of data improve the confidence in the matching of fragmentation patterns, ensuring more accurate peptide identification.
Post-Translational Modifications (PTMs) Analysis
Post-translational modifications (PTMs), such as phosphorylation, acetylation, and glycosylation, are critical for determining protein activity. Therefore, special strategies are essential for the comprehensive analysis of PTMs during de novo protein sequencing.
1. PTM Enrichment
(1) Phosphorylated proteins can be selectively enriched using techniques like TiO₂ or IMAC magnetic beads, which bind phosphopeptides with high specificity.
(2) Glycosylated proteins are typically enriched using Lectin affinity chromatography or purification via Hydrophilic Interaction Liquid Chromatography (HILIC) columns.
2. Mass Spectrometry Analysis Strategy
(1) HCD (Higher-energy C-trap dissociation) combined with EThcD (Electron Transfer Dissociation) fragmentation allows for the analysis of protein sequences while preserving post-translational modifications.
(2) Tandem mass spectrometry (MS3) further improves the precision of modification site localization by providing an additional level of fragmentation to confirm the position of PTMs.
3. Combination with Quantification Methods
(1) iTRAQ/TMT labeling techniques are widely used for quantifying PTMs, enabling the analysis of dynamic changes in protein modifications across different conditions.
(2) Stable Isotope Labeling with Amino acids in Cell culture (SILAC) is a powerful method for tracking dynamic changes in PTMs by labeling proteins with isotopically labeled amino acids.
Long Protein Sequence Assembly
1. Combining Bottom-up and Top-down Strategies
(1) Bottom-up (Analysis After Protein Degradation): This approach is suitable for analyzing complex samples, but it faces challenges in protein assembly due to the degradation of the protein into smaller peptides.
(2) Top-down (Direct Analysis of Intact Proteins): Ideal for detecting protein mutations or modifications, this method requires high-quality mass spectrometry due to its sensitivity and precision demands.
(3) Combined Strategy (Middle-down): This approach utilizes partial digestion to enhance the resolution and improve the accuracy of protein assembly.
2. Using Protein Databases to Improve Assembly Accuracy
(1) While de novo sequencing is fundamentally independent of databases, sequence alignment with resources such as Uniprot, NCBI, and others can be helpful for the following purposes:
(2) Verifying and refining partially known protein fragments, thereby increasing the confidence in the assembly.
(3) Identifying homologous proteins and predicting potential functional modification sites.
Obtaining comprehensive protein information through de novo protein sequencing necessitates the integration and optimization of several critical steps, including sample preparation, mass spectrometry analysis, data interpretation, PTM identification, and sequence assembly. This holistic approach overcomes the limitations of relying on any single technology, providing a comprehensive solution. As high-resolution mass spectrometry, single-molecule sequencing, artificial intelligence, and other advanced technologies continue to evolve, de novo sequencing will become indispensable in fields such as the exploration of unknown proteins, precision medicine, and the development of antibody-based therapies. These advancements will drive the closer integration of life sciences with the biopharmaceutical industry. MtoZ Biolabs offers de novo protein sequencing services to its clients. Our comprehensive, "one-stop" services save valuable time and effort, enabling more efficient progress in related research.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

