





Mass Spec Proteomics Service
Mass spec proteomics is a powerful approach that utilizes mass spectrometry to analyze the composition and variations of proteins in a sample, providing detailed insights into protein composition, structure, and function. Mass spectrometry has been widely applied to biological sample analysis and has evolved into an indispensable tool for proteomics research. Recently, new mass spectrometers (such as Orbitrap) and novel dissociation methods (such as electron transfer dissociation) have opened up exciting new areas of proteomic applications. Although bottom-up proteomics (analyzing proteolytic peptide mixtures) remains the mainstream approach for proteomic analysis, middle- and top-down strategies (analyzing longer peptides and intact proteins, respectively) are expected to offer a more complete characterization of protein isoforms and post-translational modifications. Mass spec proteomics relies on ionizing protein samples, fragmenting them into smaller peptides, analyzing their mass-to-charge ratios using a mass spectrometer, and matching the resulting peptide spectra to known databases. It provides information on protein quantification, characterization, identification, and post-translational modifications. This helps in understanding protein function, disease mechanisms, and biomarker discovery, making it an invaluable tool for research in areas such as drug development, personalized medicine, and cellular processes.
Service at MtoZ Biolabs
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider, provides advanced proteomics, metabolomics, and biopharmaceutical analysis services to researchers in biochemistry, biotechnology, and biopharmaceutical fields. Our ultimate aim is to provide more rapid, high-throughput, and cost-effective analysis, with exceptional data quality and minimal sample consumption. We are honored to introduce our mass spec proteomics services equipped with the Thermo Fisher Q Exactive HF and Orbitrap Fusion Lumos mass spectrometer systems, coupled with a Nano-LC system. In proteomics research, we offer three primary mass spec proteomics strategies: bottom-up, middle-down, and top-down proteomics. If you are interested in our services, please feel free to contact us, and we will be happy to assist you.
1. Bottom-Up Proteomics
Over the past few decades, mass spec proteomics has enabled precise genome-wide quantification of protein changes across multiple biological states within a single experiment. In the field of MS-based proteomics, the predominant methodology is the bottom-up strategy. This approach involves breaking down proteins into peptides through proteolytic digestion. The workflow begins with the extraction, denaturation, reduction, and alkylation of proteins from biological samples to prepare them for enzymatic digestion. The digestion is typically carried out with trypsin, which cleaves proteins at the C-terminus of lysine and arginine residues with high specificity. After digestion, peptides undergo rigorous clean-up processes, such as solid-phase extraction, to remove contaminants and, optionally, to enrich for targeted modification studies. The refined peptide mixture is then separated using high-performance liquid chromatography (HPLC), primarily reversed-phase HPLC (RPLC), based on the peptides' hydrophobicity. The separated peptides are introduced into the mass spectrometer using an electrospray ionization (ESI) source. In the mass spectrometer, the mass-to-charge ratios and signal intensities of ionized peptides and their corresponding fragmentation products are measured. Bioinformatics tools process the fragmentation spectrum data against theoretical spectra generated from in silico digestion of protein databases, with strict statistical analysis to control the false discovery rate (FDR) and ensure the reliability of results. Subsequently, the identified peptides are mapped back to their source proteins, allowing for the detailed analysis of protein composition, modifications, and interactions.
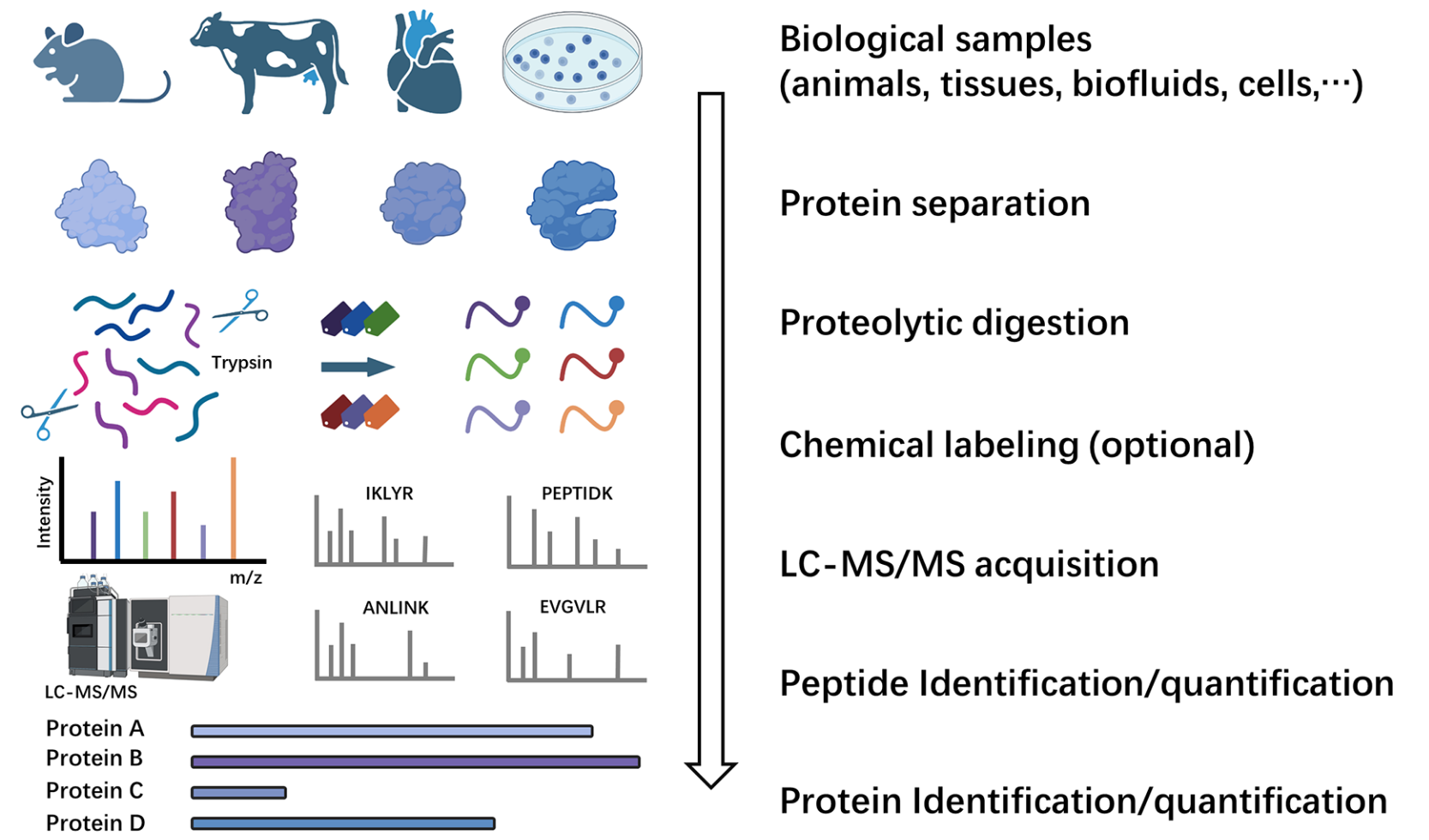
Wang, Z. et al. ACS Meas Sci Au. 2024.
Figure 1. The Workflow of Bottom-Up Proteomics
2. Middle-Down Proteomics
Middle-down proteomics involves generating and analyzing larger peptides (typically >20 amino acids), which requires the use of proteases other than trypsin. To minimize missed cleavage events, it is ideal to use proteases that cleave after a single residue. For example, endoproteinase AspN, which cleaves N-terminal to aspartate residues, and endoproteinase GluC, which cleaves C-terminal to glutamate residues, are commonly used to generate specific peptides for analysis. The middle-down approach typically avoids reversed-phase (RP) chromatography due to the poor binding and separation of larger peptides, particularly for hydrophilic proteins. Instead, weak cation exchange (WCX) hydrophilic interaction liquid chromatography (HILIC) is employed for better separation. WCX-HILIC utilizes a hydrophilic stationary phase coupled with an organic mobile phase, enabling efficient separation of larger peptides, either online or offline, for subsequent mass spectrometry analysis. So the mass spec proteomics above has become an alternative workhorse for the analysis of proteins and their modifications.
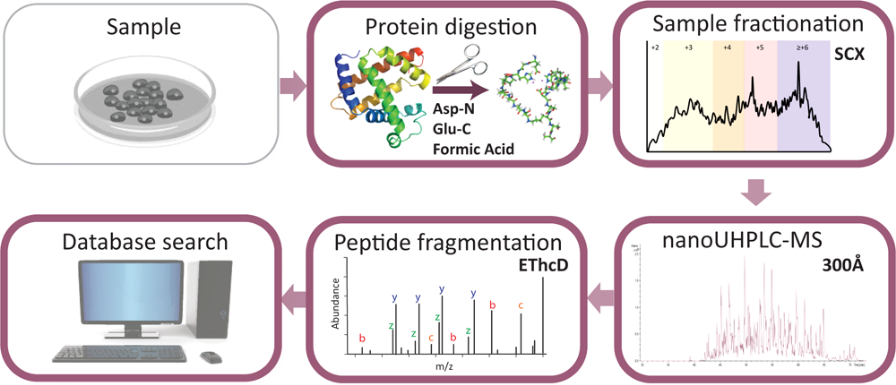
Cristobal, A. et al. Anal Chem. 2017.
Figure 2. The Workflow of Middle-Down Proteomics
3. Top-Down Proteomics
Mass spec proteomics is the most comprehensive approach for the quantitative profiling of proteins, their interactions and modifications. Top-down proteomics has become the most powerful experimental strategy for comprehensive analysis of proteoforms. The top-down approach involves three main steps: ionization to generate gas-phase ions from the protein of interest that can be transported into the mass spectrometer; full mass analysis of the ionized protein via MS1 (the upper level) and complete gas-phase fragmentation through MS2 to generate product ions that provide sequence information (the lower level); and data processing, including database searching, for the identification, characterization, and quantification of protein forms. Since top-down proteomics is applied to protein mixtures, the workflow typically requires separation of the analytes. High-quality separation capabilities are particularly important for top-down proteomics because the fragment ions generated from intact proteins result in complex spectra, where various ions with different charge states may partially overlap. Many modern mass spectrometers can reliably achieve high resolution, including Fourier transform mass spectrometry systems such as ion cyclotron resonance (FTICR) and Orbitrap instruments, as well as time-of-flight (TOF) and quadrupole-TOF (QTOF) instruments.
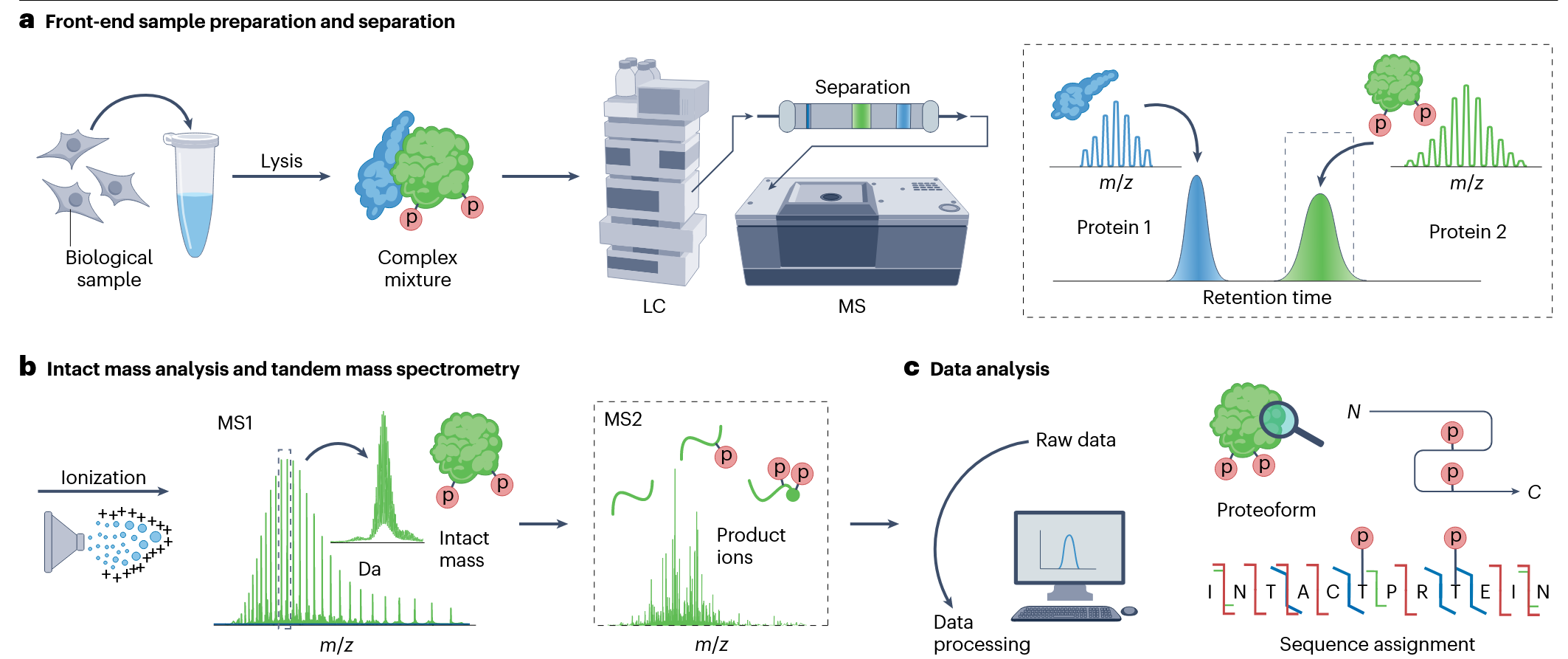
Roberts, DS. et al. Nat Rev Methods Primers. 2024.
Figure 3. The Workflow of Top-Down Proteomics
Service Advantages
1. MtoZ Biolabs utilize advanced LC-MS/MS platforms, including the Thermo Fisher Orbitrap Fusion Lumos, ensuring high resolution and reliable data acquisition.
2. Our mass spec proteomics offers unparalleled sensitivity and precision, enabling the identification and quantification of proteins in complex biological matrices, providing accurate results for your research.
3. MtoZ Biolabs provides comprehensive data analysis using cutting-edge bioinformatics tools, delivering actionable insights tailored to diverse research needs.
4. Our experienced scientific team offers end-to-end support, from experimental design to data interpretation, ensuring a seamless and efficient workflow.
Applications
Mass spec proteomics can be ued in:
1. Biomarker Validation
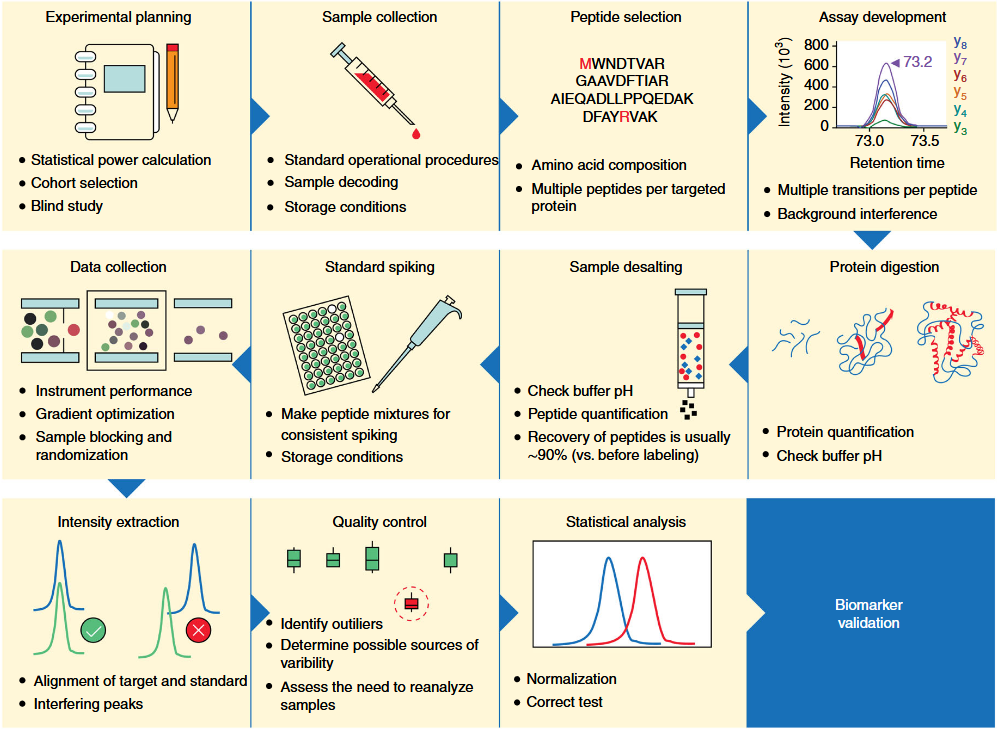
Nakayasu, ES. et al. Nat Protoc. 2021.
2. Disease Research
Birhanu, AG. et al. Clin Proteomics. 2023.
FAQ
Q1: What should be done if the mass spec proteomics shows contamination in the protein sample during protein identification?
Answer: When protein mass spec proteomics shows sample contamination, the following measures can be taken to address the issue:
1. Identify the Source of Contamination
First, it is important to determine the source of the contamination, which could come from the experimental environment, equipment, reagents, or the sample itself. This information can be obtained through comparison experiments, such as using a blank control group or changing experimental conditions and materials.
2. Improve Sample Handling
If the contamination originates from the sample, improvements in sample handling can help reduce it. For example, optimizing sample extraction, purification, and storage methods can minimize the introduction or generation of contaminants.
3. Improve Experimental Conditions and Equipment
If the contamination is from experimental conditions or equipment, such as the ion source, consider replacing or cleaning the equipment, or changing the reagents and conditions used.
4. Optimize Data Processing
During the data processing stage, more accurate data processing software and methods can be used to identify and remove contaminants. For example, using mass spectrometry databases that can detect contaminants or employing mass spectrometry pairing algorithms that reduce false identification can help.
These four steps provide a basic approach to handling sample contamination in mass spec proteomics, and specific implementation may need to be adjusted and optimized based on the specifics of the experiment.
Deliverables
1. Comprehensive Experimental Details
2. Materials, Instruments, and Methods
3. Relevant Liquid Chromatography and Mass Spectrometry Parameters
4. The Detailed Information of Proteomics
5. Mass Spectrometry Image
6. Raw Data
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

