





Mass Spectrometry for Proteins Analysis Service
Mass Spectrometry for Proteins Analysis Service utilizes mass spectrometry (MS) technology to analyze proteins in terms of identification, quantification, post-translational modifications (PTMs) analysis, interaction studies, and structural characterization. Proteins are the key executors of biological functions, and their functional state, interactions, and modifications directly influence cellular physiological processes. Traditional protein analysis methods such as Western Blot and ELISA provide qualitative or semi-quantitative information but have limitations in high-throughput capacity, comprehensiveness, and sensitivity. The advent of mass spectrometry has changed protein analysis by enabling precise characterization of protein structure, modifications, and dynamic changes. Mass Spectrometry for Proteins Analysis can detect proteins at the fmol level and simultaneously analyze both high-abundance and low-abundance proteins, covering thousands of proteins in a single run to achieve high-throughput analysis. Leveraging an advanced mass spectrometry platform, MtoZ Biolabs provides Mass Spectrometry for Proteins Analysis Service that is suitable for a wide range of applications, including disease biomarker discovery, drug target research, and signaling pathway analysis, supporting cutting-edge life science research.
Analysis Workflow
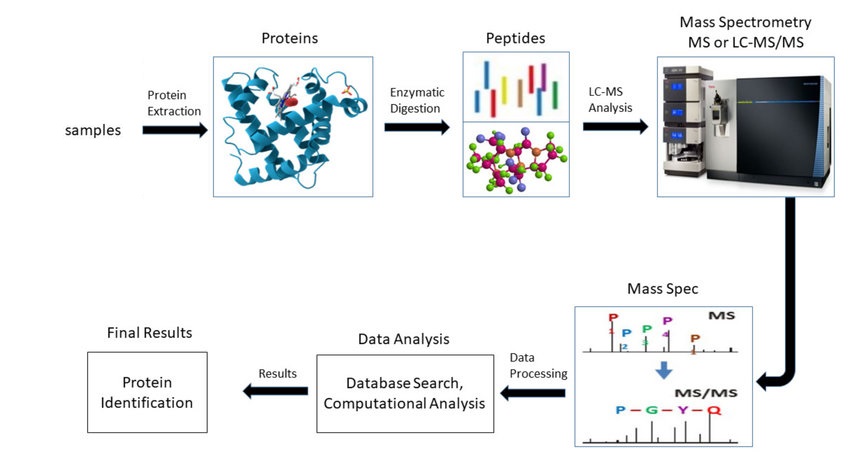
Khan M Z I. et al. Cells. 2022.
The main workflow of Mass Spectrometry for Proteins Analysis Service includes:
1. Sample Preparation:
Protein extraction, purification, and digestion. For post-translational modification (PTM) analysis, enrichment of the target modified peptides is required.
2. Mass Spectrometry Analysis
Proteins or peptides are ionized using electrospray ionization (ESI) or matrix-assisted laser desorption/ionization (MALDI) technology. Tandem mass spectrometry (MS/MS) is then used to fragment and detect the proteins or peptides.
3. Data Analysis
Combined with software such as SEQUEST, Mascot, and MaxQuant, the fragment ion spectra were compared with databases (such as Uniprot and Swiss-Prot) to confirm the protein identity. For quantitative analysis, label-free quantification (LFQ) or labeling techniques such as TMT, iTRAQ are used to determine changes in protein abundance. If the study involves PTMs, specific modification sites are identified and analyzed. Additionally, bioinformatics tools such as Gene Ontology (GO) and KEGG pathway analysis can be used to further interpret protein functions, study protein interactions, and explore signaling pathways.
Service Advantages
1. Advanced Analysis Platform: MtoZ Biolabs established an advanced Mass Spectrometry for Proteins Analysis Service platform, guaranteeing reliable, fast, and highly accurate analysis service.
2. One-Time-Charge: Our pricing is transparent, no hidden fees or additional costs.
3. High-Data-Quality: Deep data coverage with strict data quality control. AI-powered bioinformatics platform integrates all Mass Spectrometry for Proteins Analysis Service data, providing clients with a comprehensive data report.
Applications
1. Protein Identification
Analysis of protein composition in complex biological samples such as serum, tissues, and cell lysates.
Characterization of protein mutations, splice variants, and post-translational modifications (PTMs).
2. Protein Quantification Analysis
Disease Biomarker Screening: Comparing protein expression levels under different physiological or pathological conditions.
Drug Target Analysis: Investigating drug mechanisms and their regulatory effects on protein levels.
3. Post-Translational Modifications (PTMs) Analysis
Phosphorylation: Studying kinase/phosphatase regulatory mechanisms in signaling pathways.
Glycosylation: Identifying glycoprotein modifications and analyzing their roles in protein stability and function.
Acetylation, Ubiquitination: Investigating protein degradation and epigenetic modifications.
4. Protein Interaction Analysis
Co-IP-MS (Co-Immunoprecipitation Mass Spectrometry): Identifying protein complex components.
Pull-Down-MS: Investigating protein-ligand or protein-nucleic acid interactions.
Crosslinking-MS: Provide spatial structural information of protein interactions.
5. Protein Structural Analysis
Native MS: Studying protein complexes and conformational states.
HDX-MS (Hydrogen-Deuterium Exchange Mass Spectrometry): Analyzing protein structural dynamics and protein-protein interaction interfaces.
FAQ
Q. How to Select the Appropriate Mass Spectrometry Acquisition Mode (DDA, DIA, MRM, PRM)?
Choosing the appropriate mass spectrometry analysis mode depends on the research objective, sample complexity, and data requirements:
1. DDA (Data-Dependent Acquisition)
Application: Protein identification and proteomics research.
Advantages: Enables identification of unknown proteins with high data quality.
Disadvantages: Randomized selection favors high-abundance proteins, potentially missing low-abundance proteins.
2. DIA (Data-Independent Acquisition)
Application: Large-scale protein quantification and multi-sample comparisons.
Advantages: Comprehensive coverage of all detectable peptides, improving the detection of low-abundance proteins.
Disadvantages: Complex data requiring advanced computational analysis.
3. MRM (Multiple Reaction Monitoring)
Application: Targeted protein quantification, biomarker validation, and clinical diagnostics.
Advantages: High specificity and sensitivity, suitable for large-scale sample analysis.
Disadvantages: Limited to known target proteins, unsuitable for whole-proteome screening.
4. PRM (Parallel Reaction Monitoring)
Application: High-precision targeted protein quantification, ideal for validation studies.
Advantages: More flexible than MRM, compatible with high-resolution mass spectrometry (e.g., Orbitrap), providing higher analytical precision.
Disadvantages: Lower throughput compared to MRM, making it less suitable for large-scale clinical applications.
In summary, DDA is recommended for unknown protein identification, DIA for high-coverage proteome quantification, and MRM/PRM for precise quantification of specific proteins. MtoZ Biolabs offers end-to-end mass spectrometry based protein analysis service, including experimental design. Contact us for more details.
Deliverables
1. Comprehensive Experimental Details
2. Materials, Instruments, and Methods
3. Total Ion Chromatogram & Quality Control Assessment (project-dependent)
4. Data Analysis, Preprocessing, and Estimation (project-dependent)
5. Bioinformatics Analysis
6. Raw Data Files
Case Study
This study utilized Mass Spectrometry for Proteins Analysis to identify proteomic changes regulated by USP14 and to examine alterations in the ubiquitination status of α-synuclein. The results revealed that inhibition of USP14 promotes α-synuclein degradation, reducing its accumulation in dopaminergic cells, thereby influencing the pathological processes associated with Parkinson’s disease (PD).
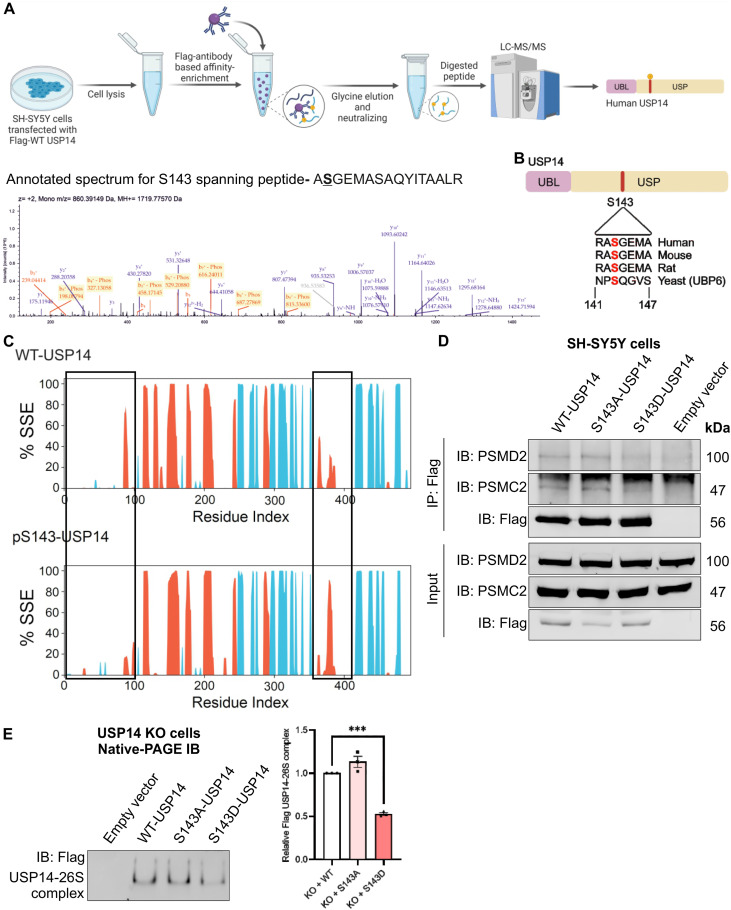
Srinivasan V. et al. Heliyon. 2025.
How to order?

