





Quantitative Proteomics Analysis Service | Biopharmaceutical
Due to significant developments in mass spectrometry (MS) technology and software, quantitative proteomic analysis has become the primary method for protein quantification. This technique is mainly based on bottom-up (shotgun) proteomics, which relies on the analysis of proteins digested by proteases in biological samples. Depending on the study, optional enrichment of organelles, modifications, or affinity purification should be performed beforehand. For instance, proteomic study of expressed proteins can be conducted on whole-cell lysates or purified organelles, typically involving the specific enrichment of peptides with post-translational modifications (PTMs), and affinity purification of interacting partners. Pre-separation at the protein or peptide level can be introduced to increase coverage and dynamic range. Subsequently, the use of chromatography and MS for analysis generates large MS datasets and tandem mass spectrometry (MS/MS) data, which include quantitative peptide information and characteristic fragment patterns for sequence determination.
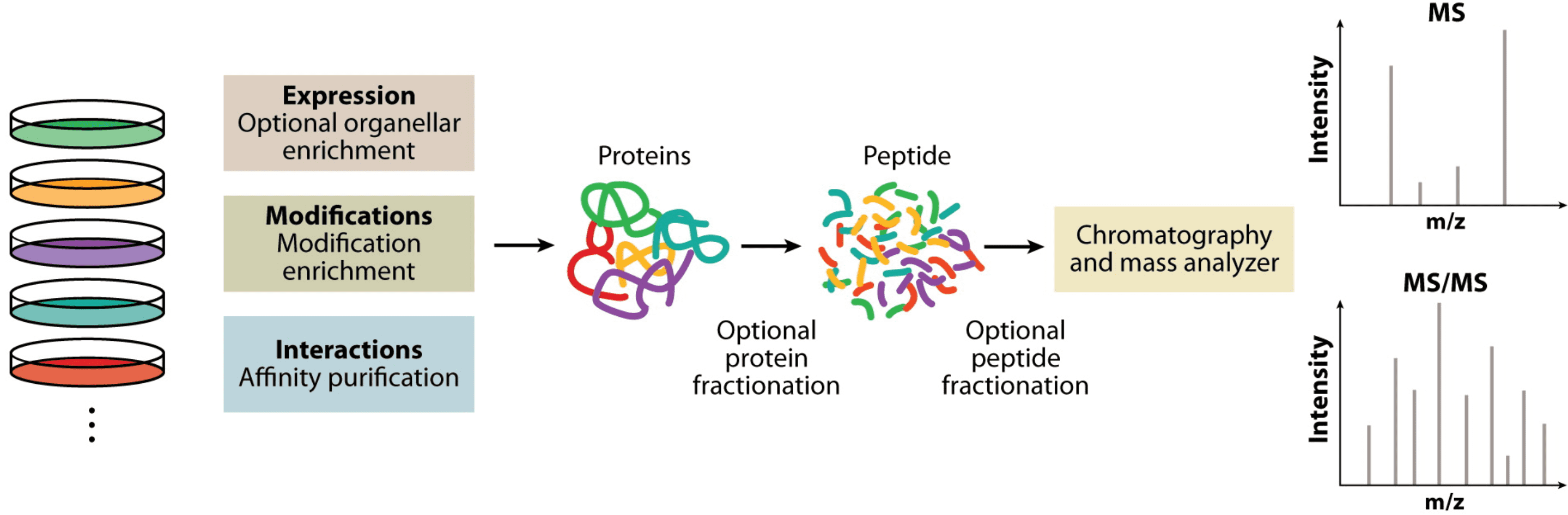
Figure 1. Overview of the Bottom-Up Proteomics Workflow[1]
Due to the limitations of spectral counting methods for protein quantification, chemical and metabolic labeling methods for relative and absolute protein quantification were developed in the late 1990s. Labeling techniques include isotope-coded affinity tags (ICAT), stable isotope labeling with amino acids (SILAC) in cell culture, tandem mass tags (TMT), and isobaric tags for relative and absolute quantitation (iTRAQ). Labeling methods offer higher accuracy and precision. But it has limitations in multi-step sample preparation, costs more, and uses more samples than other methods. Recent methods such as absolute quantification (AQUA), multiple reaction monitoring (MRM), quantification concatemers (QconCATs), protein standard absolute quantification (PSAQ), and stable isotope standards and capture by anti-peptide antibodies (SISCAPA) address some of these limitations.
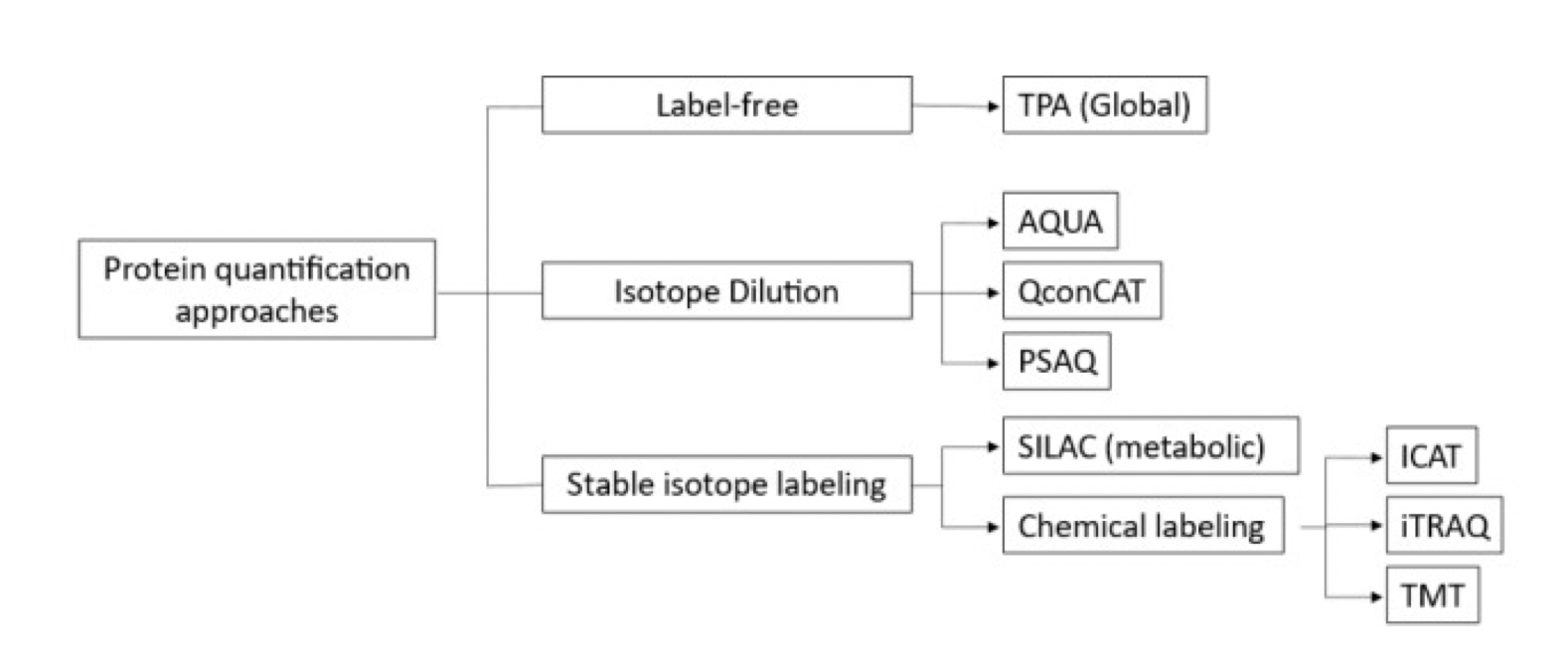
Figure 2. Common Quantitative Proteomic Labeling Strategies[2]
As proteomics continues to evolve, its application are becoming increasingly widespread, playing a significant role in finding diagnostic markers for diseases, screening drug targets, and mechanism research. hence it has been extensively applied in medical research, which include following aspects: (1) Diagnosis: By using high-throughput methods to screen and identify disease-specific biomarkers, important evidence for disease diagnosis, especially early diagnosis, can be provided; (2) Therapeutics: Using proteomics to study the interactions between proteins in healthy or diseased organisms, the pathological basis of diseases. Proteomics can identify potential drug targets, examine the druggability of selected protein targets, design and develop new drugs against candidate therapeutic protein targets and identify drug components, toxicity, and mechanisms of action to improve drug efficacy and safety; (3) Prevention/Prognosis: Identifying new proteins with high tumor-specific immunological activity aids in the research and development of anti-tumor vaccines; proteomics research provides information related to primary tumors and their metastasis, which is significant for tumor prognosis analysis; (4) Mechanism research: Finding differential proteins to identify proteins related to diseases. And further studies on the roles abnormal proteins playing in the disease development can be conducted to help reveal the pathogenesis of diseases at the molecular level.
MtoZ Biolabs has a comprehensive proteomics analysis platform, mainly consisting of various mass spectrometers and related auxiliary equipment (such as high-performance liquid chromatography), and the platform has complete data storage, data retrieval, and data analysis capabilities. The services offered by the platform include multiple-source protein extraction, mass spectrometry sample pretreatment, various chemical/metabolic labeling, high-performance liquid chromatography sample fractionation, and mass spectrometry data collection, enabling research in areas such as quantitative proteomics, phosphoproteomics, etc. The team can explore the best experimental conditions through multi-gradient experiments, collect data through mass spectrometry, and analyze changes in protein expression and modifications through bioinformatics. MtoZ Biolabs can provide services like protein extraction, sample preparation, chemical/metabolic labeling, mass spectrometry data collection, and experimental data analysis, offering a one-stop solution to quantitative proteomics issues.
References
[1] Cox J, Mann M. Quantitative, high-resolution proteomics for data-driven systems biology. Annu Rev Biochem. 2011;80:273-99. doi: 10.1146/annurev-biochem-061308-093216. PMID: 21548781.
[2] Ahire D, Kruger L, Sharma S, Mettu VS, Basit A, Prasad B. Quantitative Proteomics in Translational Absorption, Distribution, Metabolism, and Excretion and Precision Medicine. Pharmacol Rev. 2022 Jul;74(3):769-796. doi: 10.1124/pharmrev.121.000449. PMID: 35738681; PMCID: PMC9553121.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
Label-Free DIA Quantitative Proteomics
Quantitative Phosphoproteomics Service
iTRAQ-based Quantitative Proteomics Analysis
How to order?

