





Small Molecule Drug Screening Service-Enzymes
1. Kinase Inhibitors
Kinases, in simple terms, are enzymes that transfer the phosphate groups of ATP to substrates. Kinases catalyze the transfer of γ-phosphate groups of ATP to substrates, mediate most signal transduction, and regulate a variety of cellular activities, including proliferation, survival, apoptosis, metabolism, transcription, differentiation, and a wide range of other cellular processes. Accumulated pharmacological and pathological evidence suggest that kinases are promising drug targets for the treatment of a variety of diseases, such as cancer, inflammatory diseases, central nervous system (CNS) diseases, cardiovascular diseases, and complications of diabetes.
The targets of small molecule inhibitors targeting kinases mainly include tyrosine kinases (TKs) and multiple serine/threonine kinase families. (1) AGC: Protein kinase A, G, C: These kinases are serine/threonine kinases, which are involved in many key intracellular signal transduction pathways. (2) CMGC: CMGC is named after the initials of four kinases: CDK, MAPK, GSK3, and CLK, but this group is not limited to these four subgroups, with a total of nine subgroups. The kinases in this group are all serine/threonine kinases, which are widely involved in the regulation of cell cycle, growth, differentiation, splicing, metabolism, etc. (3) CAMK (Ca2+/calmodulin-dependent protein kinase): the kinases in this group are all serine/threonine kinases. Calmodulin (CaM) binds to Ca2+ and binds to the C-terminus CBD domain of CAMK, thereby opening the AID domain and releasing the N-terminal catalytic domain for kinase activation. (4) CK1 (Casein kinase 1/cell kinase 1): type I casein kinase family, which is a serine/threonine kinase, which can be divided into three subgroups. (5) STE: all serine/threonine kinases. It is mainly divided into three families: STE7, STE11, and STE20, which are the key nodes of MAPK signaling pathway. (6) TK : The kinases in this group are all TKs, more than half of the members are cell membrane receptors (and receptor tyrosine kinase, RTK), and the other members mostly play functions near the cell membrane. (7) TKL (Tyrosine Kinase-Like): very similar to TK, some members are TKs, but many members are serine/threonine kinases, and this group of kinases can be divided into 8 families. (8) Atypical kinases: This family of protein kinases has no structural similarity with the above 8 groups of kinases (also known as ePK, eukaryotic protein kinases). (9) RGC (Receptor Guanylate Cyclases): This group contains a number of guanylate cyclase receptors with a guanylate cyclase region (which converts GTP to cGMP as a second messenger) and an inactivated kinase domain. (10) PKL (Protein Kinase-Like): includes a series of lipids, sugars, and other small molecule kinases.
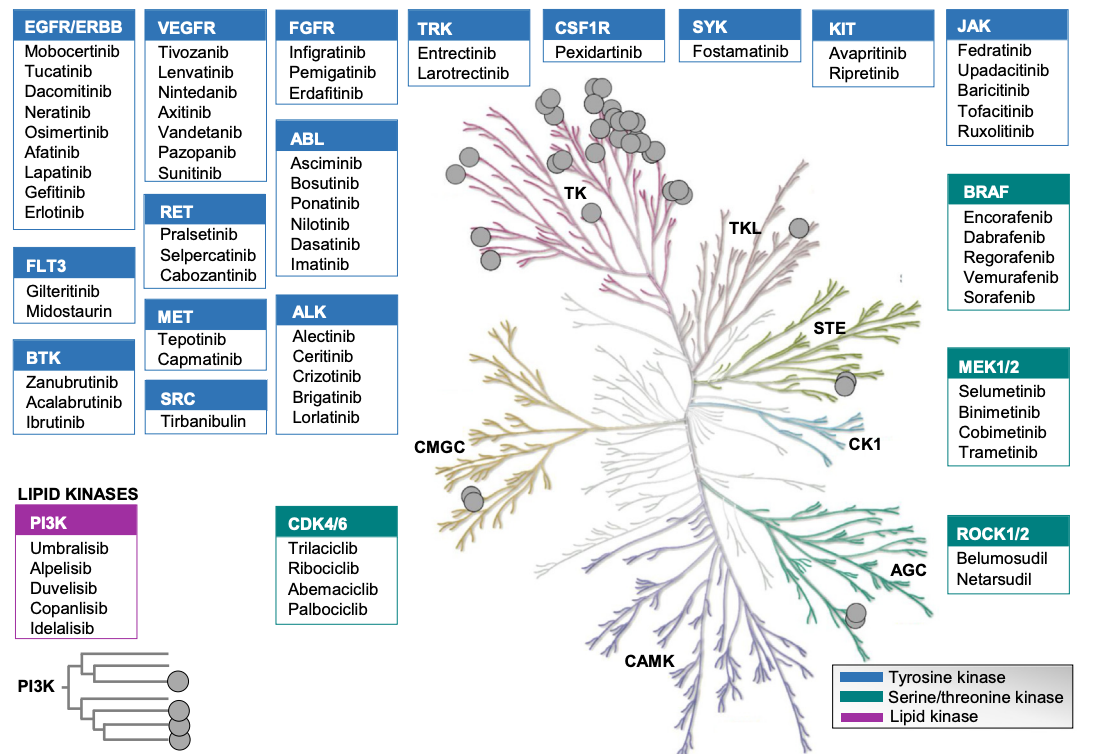
Figure 1. Summary of Small Molecule Targets Targeting Kinases [6]
Beyond the traditional type I and II inhibitors that bind to the ATP-binding pocket, an increasing number of inhibitors have been reported as allosteric type III and IV inhibitors, covalent inhibitors, and bivalent inhibitors, as well as new chemistries, including molecular gels, proteolytic-targeted chimeras (PROTACs), and other types of kinase-targeted, proximity-induced bifunctional small molecules. Among them, the type I inhibitor binds to the active kinase conformation, and the DFG motif points to the ATP-binding pocket. Type II inhibitors conformationally bind to inactive kinases, with DFG motifs pointing to the outside of the ATP-binding pocket. Type III and type IV inhibitors bind to allosteric pockets adjacent to or distal to the ATP binding pocket, respectively. Covalent inhibitors containing electrophilic warheads form reversible or irreversible covalent bonds with nucleophilic amino acid residues of kinase substrates, which in turn inhibit kinase function. Bifunctional degraders-PROTACs can recruit E3 and E2 ubiquitin ligases to add ubiquitin to target kinases for ubiquitin-mediated proteasomal degradation.
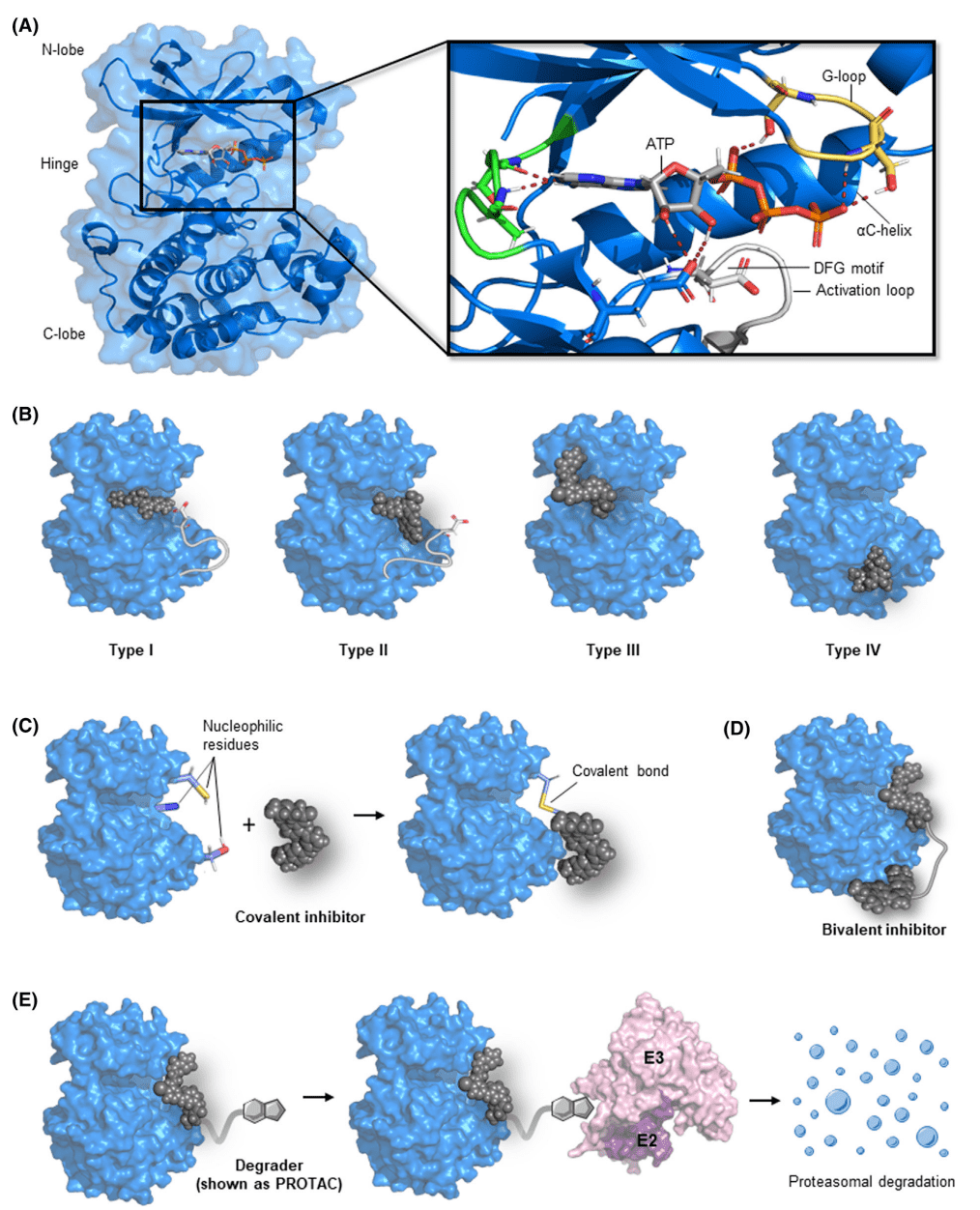
Figure 2. Types of Small Molecule Inhibitors that Target Kinases[6]
In recent years, an array of screening techniques for drug discovery have been developed. In particular, many HTS methods that are compatible with inhibitor screening have made great progress, and these methods can be used to quickly and simply identify libraries containing thousands or millions of compounds. Typically, these HTS methods use fluorescence or chemiluminescence to detect changes in signal intensity. These methods all use microplates as carriers, which have the advantages of low reagent consumption, high detection speed and high throughput, and have become some of the most commonly used protein kinase detection methods. According to the difference of detection targets, it can be divided into detection of ATP consumption, detection of ADP production, detection of peptide substrates, and detection of kinase itself.
(1) Detect the Amount of ATP Remaining
Some methods can measure enzyme activity by measuring the amount of ATP that is not consumed in a kinase reaction, such as Promega's Kinase-Glo™ Technology. This method uses the ATP remaining after the kinase reaction to convert fluorescein into oxidized luciferin and emits chemically cold light, and the fluorescence signal value is inversely proportional to the kinase activity.
(2) Detect the Amount of ADP Generated
This type of method makes it easier to detect the kinase activity of some non-peptide substrates, such as lipokinase, and can also measure the activity of some ATPases. These methods can be divided into ADP conjugation reactions, i.e., the conversion of ADP into other substances for detection, or the detection of ADP antibodies.
(3) Fluorescence Polarization (FP)
FP is a method that can be used to quantitatively measure the intensity of protein-protein interactions. FP is based on the relationship between the polarization of the fluorophore and the modulation of its anisotropy value. Polarized light is generated by passing light through an excitation polarization filter, which excites the fluorophore, and then depolarizes it through the molecular rotation portion of the fluorophore. The degree of polarization parallel to or perpendicular to the excitation plane can be monitored by an emission polarizer. The intensity of the emitted light is determined by the size of the fluorophore. When studying PPI inhibitors using this method, a fluorescently labeled peptide containing a "hot spot" of one of the protein chaperones is typically used as a fluorophore. When the fluorescently labeled peptide is freely present in solution, it rapidly rotates and depolarizes the initially polarized light, resulting in a low FP signal.
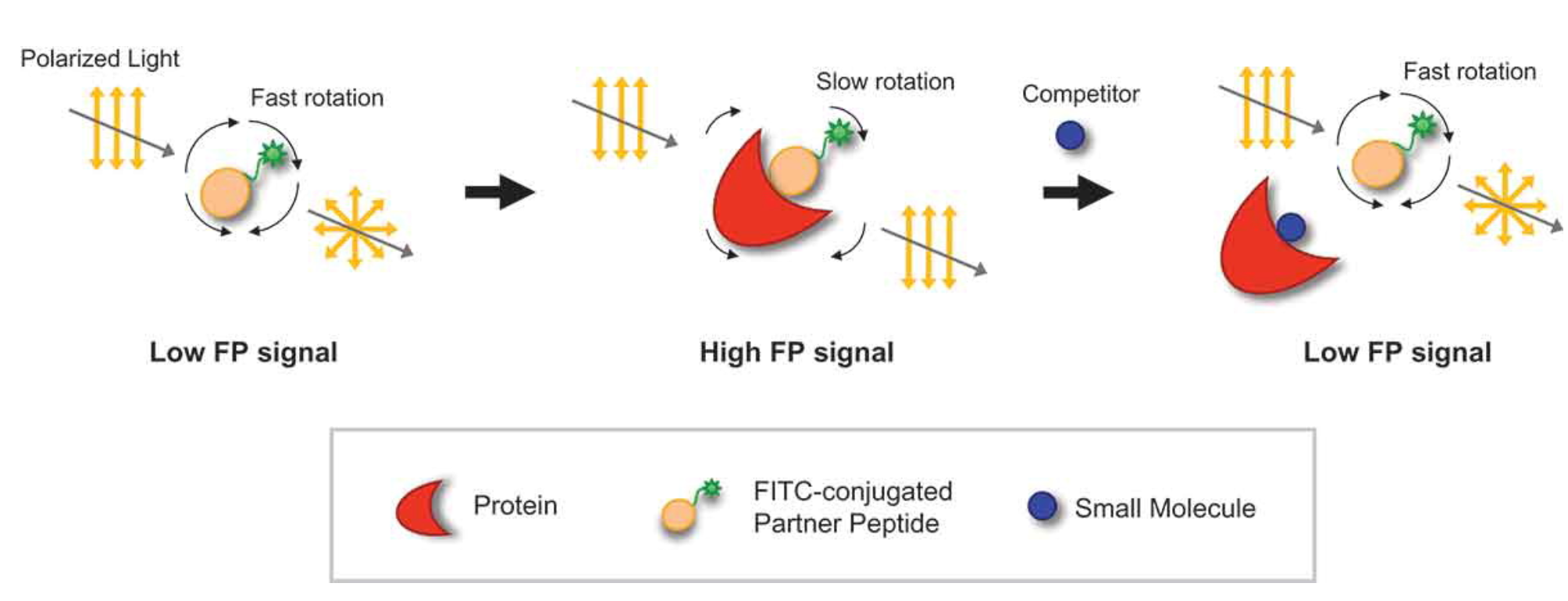
Figure 3. Principle of Fluorescence Polarization
(4) Fluorescence Resonance Energy Transfer (FRET)
FRET is a method of measuring the energy transfer between two fluorophores that are sensitive to light. When the electron-excited donor fluorophore is close to the acceptor molecule, the energy is transferred and absorbed into the acceptor fluorophore. Energy transfer occurs only when the distance between the donor and acceptor is less than 10 nm, depending on the type of acceptor and the arrangement of fluorescent molecules. Fluorescence-conjugated energy transfer is a fully automated method for kinase activity assays. This method links the "acceptor" fluorescent molecule to the "donor" fluorescent molecule, and when the "donor" fluorescent molecule is excited, the non-radioactive energy is transferred to the "acceptor" fluorescent molecule, and a fluorescent signal is generated.
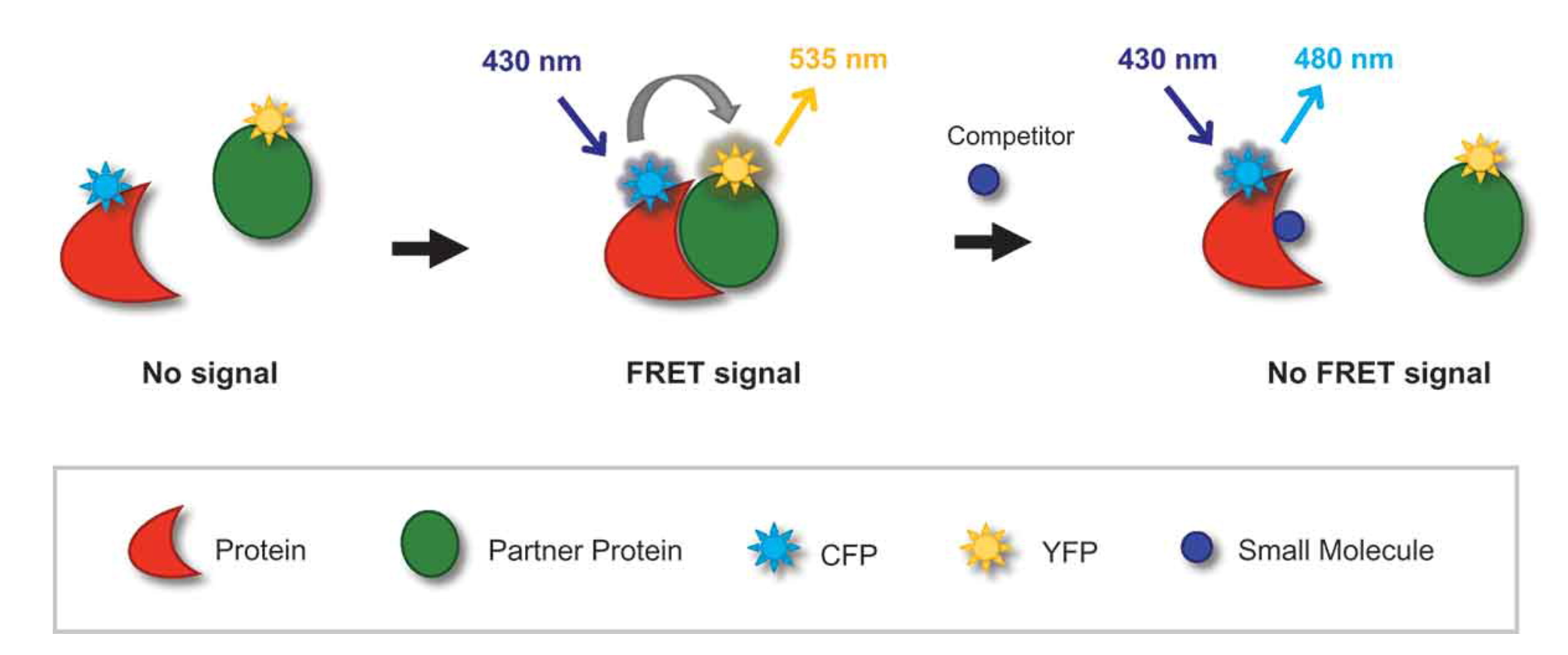
Figure 4. Principle of FRET
(5) Time-Resolved Fluorescence Resonance Energy Transfer (TR-FRET)
TR-FRET uses fluorescent molecules with a long decay time, such as europium (Eu), samarium (II), terbium (Tb) and other lanthanides, and detects the fluorescence values after a period of excitation. TR-FRET often requires the use of monoclonal antibodies (mAbs) that bind to phosphorylated peptide substrates.
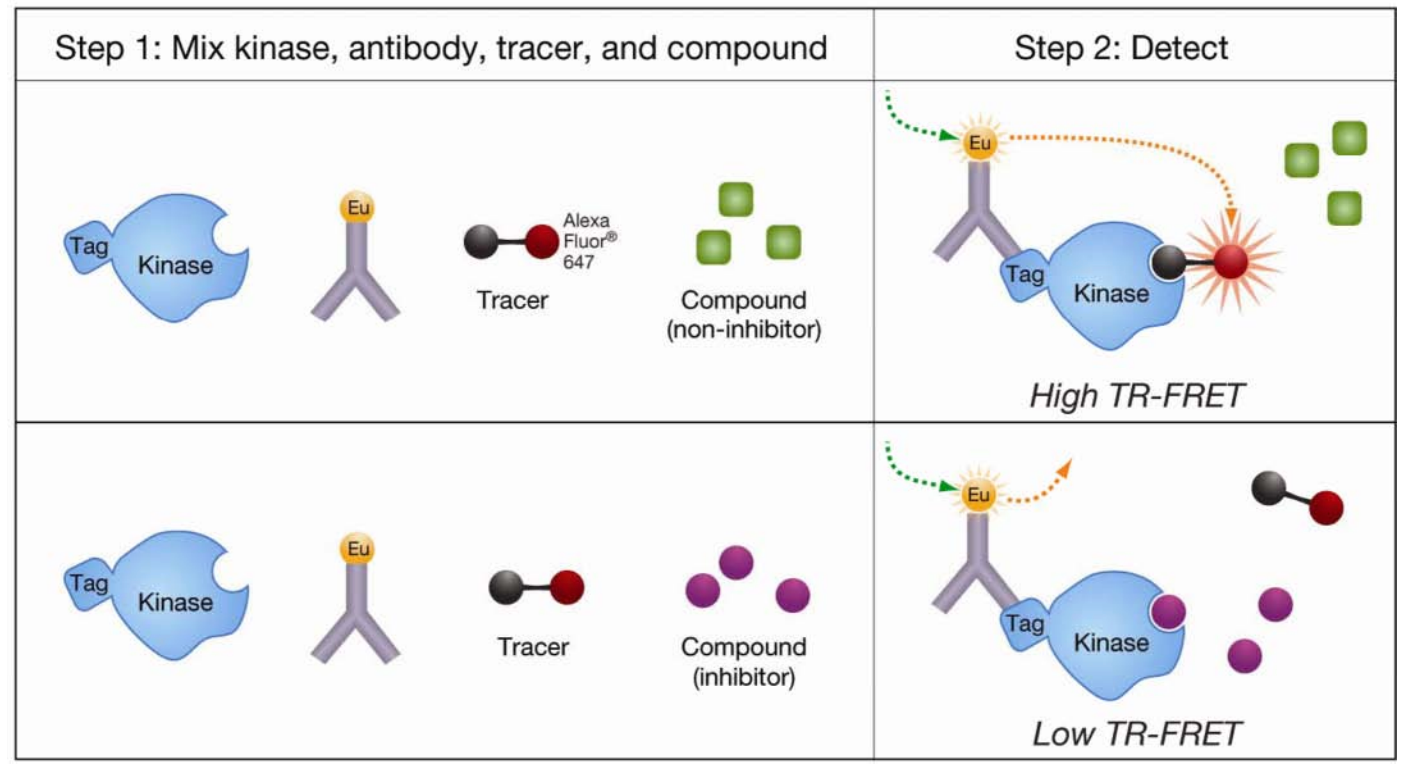
Figure 5. Principle of TR-FRET [7]
(6) Migration Change Method
After the kinase reaction, part of the substrate is added to the phosphate group by the kinase to become the product, so there is a difference of 3 charges between the substrate and the product. The mobility-altering method takes advantage of this difference and uses capillary electrophoresis to separate the substrate and product.
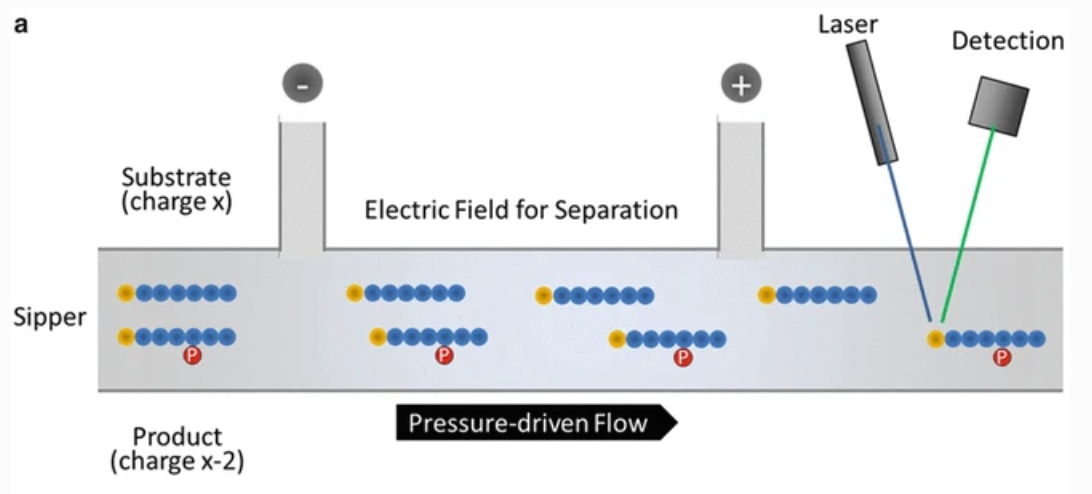
Figure 6. The Principle of Migration Rate Change Method [8]
(7) Ligand Kinase Binding Assays
In HTS and kinase zymogram screening, the use of kinase activity assays is overwhelmingly dominant. It can also be used to optimize the structure of a lead compound by monitoring kinase and substrate binding, especially when the basal activity of the purified kinase is low or its biochemical activity is unknown.
In addition to kinases, small molecule inhibitor targets targeting enzymes include: (1) Protein phosphatases (PPs) are enzymes present in the body that dephosphorylate proteins or substrates. PPs are broadly divided into three categories based on the specific amino acid species they dephosphorylate: protein serine/threonine phosphatases and protein tyrosine phosphatases, which also include bispecific phosphatases. DiFMUP can be used as a substrate to hydrolyze the phosphate group under the action of phosphatase, release the fluorophore, and detect the fluorescence value at 360/460, thus characterizing the inhibition effect of small molecule compounds. (2) Phosphodiesterase (PDEs) can hydrolyze the second intracellular messenger (cAMP, cyclic adenosine monophosphate or cGMP, cyclic guanosine monophosphate), cAMP and cGMP act as the second messenger of neurotransmitters, hormones, light and odor and other substances, and widely act on intracellular target organs. (3) Epigenetics refers to the fact that the DNA sequence does not change, but the gene expression is heritable. This alteration can be stably passed on to offspring during the development of organisms and the proliferation of cells. The main molecular mechanisms are DNA methylation, non-coding RNA regulation, histone modification, chromatin remodeling, etc. The detection method can be selected according to the biological mechanism of action of each type of protein.
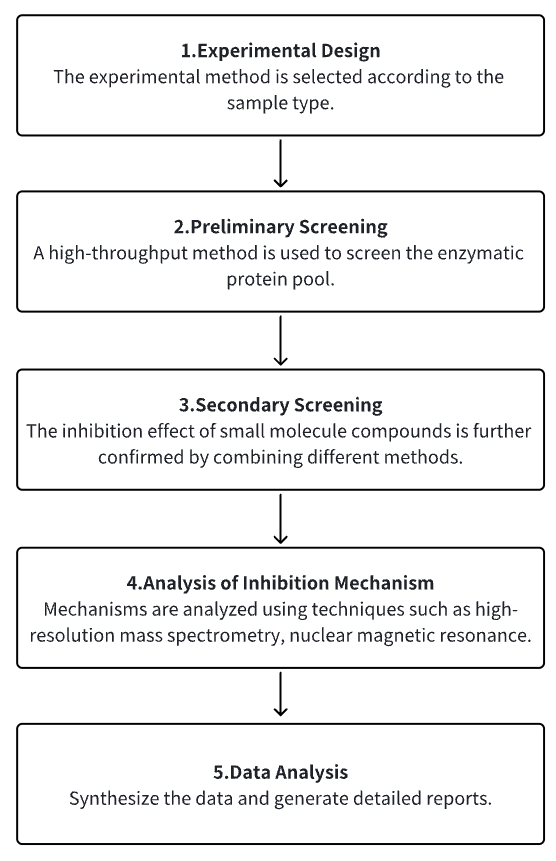
Analysis Workflow
1. Choose the Experimental Method Based on the Specific Requirements of the Experiment
2. Preliminary Screening of Enzyme Protein Library
3. Secondary Screening Validation of Small Molecules with Research Potential
4. Analysis of Small Molecule Inhibition Mechanism
5. Data Analysis
Service Advantages
1. Multiple Enzyme Protein Libraries (kinases, phosphatases, etc.)
2. Validation of Various Experimental Methods (thermal drift detection, TR-FRET analysis, Biacore detection of biological macromolecule interactions, etc.)
3. High Confidence, High-Precision Mass Spectrometry, and Other Technical Analysis Mechanisms
4. Comprehensive Bioinformatics Analysis
Sample Results
1. FRET Biosensor-Based Screening of ERK and AKT Active Kinase Inhibitors Reveals Differential Kinase-Dependent Proliferation of TNBC Cells [9]
Enhancing protein kinase expression and activity is essential for tumor cell proliferation and cancer progression. These different cancer-associated kinases form a complex network of interdependent signaling. Assessing the effects of various kinase inhibitors on these networks is critical to understanding the efficacy of kinase inhibitors in cancer therapy. Dynamic activation of some kinases can be monitored by FRET biosensors with high temporal resolution. A high-throughput imaging method based on FRET biosensors has been established to determine ERK and AKT activity in two triple-negative breast cancer (TNBC) cell lines HCC1806 and Hs578T. The study evaluated the effect of a kinase inhibitor repertoire containing > 350 different kinase inhibitors (KI) on ERK and AKT kinase activity using the FRET HTS setting.
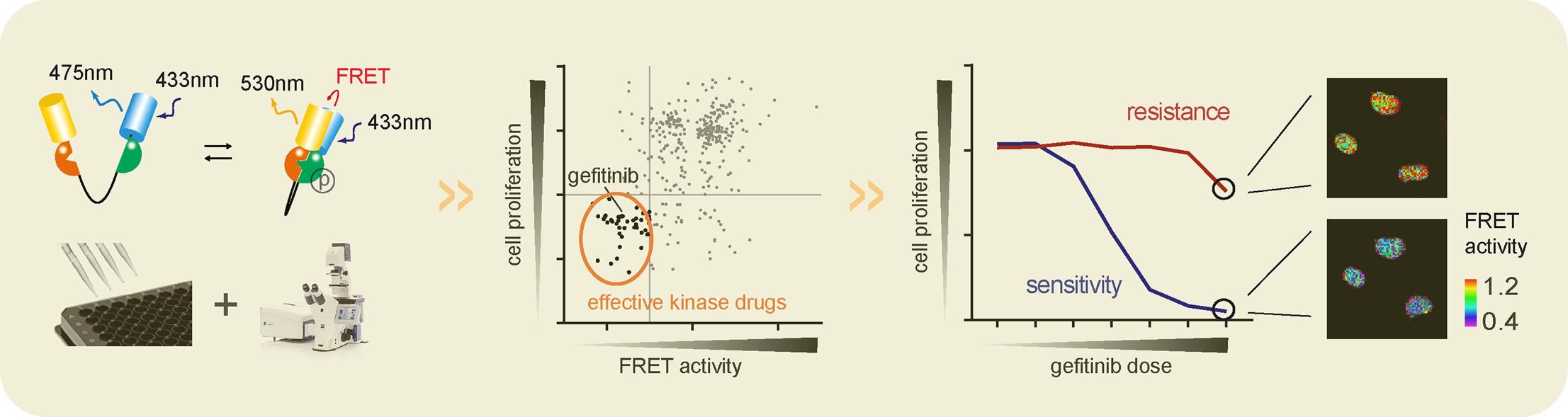
Figure 7. FRET Biosensor-Based Screening of ERK and AKT Active Kinase Inhibitors
2. Virtual Screening Combined with High-Throughput Screening to Identify Novel Rho Kinase I (ROCK-I) Inhibitors [10]
Rho kinase (ROCK) inhibitors are effective candidates for the treatment of neurological and cardiovascular diseases. Here, we report a novel class of ROCK-I inhibitors discovered through a combination of virtual screening and high-throughput screening methods. A database of 12,280 compounds was screened using a pharmacophore model based on common characteristics of known representative ROCK inhibitors. We then applied the LigandFit model to reduce the number of hits. A high-throughput screening model based on the kinase-Glo luminescent kinase assay was established to identify inhibitors observed in the virtual screening model.
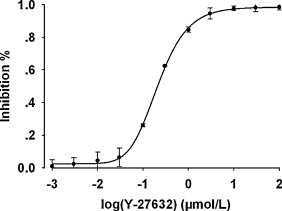
Figure 8. The Activity of Recombinant Human ROCK-I Inhibited by Y-27632 as Screened
3. Mechanistic Characterization of Tyrosine Phosphatase Inhibitors [11]
Celastrol is a natural pentacyclic triterpene used in traditional Chinese medicine (TCM) to have a significant weight loss effect. Celastrol mice administered at 100 μg/kg were observed a reduction of food consumption and body weight through leptin-dependent mechanisms, but their targets in this pathway remain elusive. In vivo studies have demonstrated that celastrol-induced weight loss is primarily mediated by the inhibition of leptin-negative regulatory protein tyrosine phosphatase (PTP) 1B (PTP1B) and T cell PTP (TCPTP) in the arcuate nucleus (ARC) of the hypothalamus. In vitro, Celastrol has been shown to reversibly bind to and inhibit non-competitive PTP1B and TCPTP. The NMR data mapped the binding site to an allosteric site in the catalytic domain near the active site. By using a panel of PTPs involved in hypothalamic leptin signaling, the results suggest that Celastrol also inhibits PTEN and SHP2, but is inactive against other phosphatases of the PTP family.
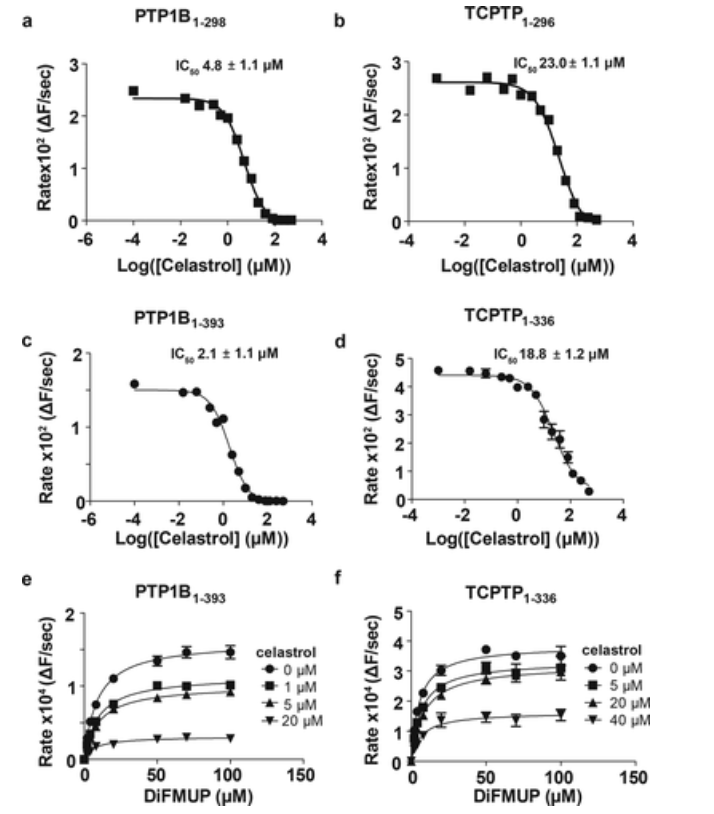
Figure 9. Enzyme Kinetics Study of Celastrol Inhibition of PTP1B and TCPTP
Sample Submission Requirements
1. Please provide processed and purified samples whenever feasible.
2. Try to avoid contamination by impurities.
Services at MtoZ Biolabs
1. Complete experimental procedures.
2. Relevant instrument parameters.
3. Raw mass spectrometry data.
4. Detailed information on small molecule inhibitor screening (including inhibition ranking, IC50 inhibition of enzyme activity, reversibility of inhibition effect, selectivity of similar protein inhibition, binding site of inhibition of enzyme activity, etc.).
Applications
1. Development of New Methods for Drug Screening
Studies have discovered novel Janus kinase 1 (JAK1) inhibitors through a combination of machine learning, structure-based pharmacophore modeling, and biological evaluation. JAK1 plays a key role in most cytokine-mediated inflammation, autoimmune responses, and various cancers through the JAK/STAT signaling pathway. Therefore, inhibition of JAK1 is an attractive therapeutic strategy for several diseases. Recently, high-performance machine learning techniques have been increasingly applied to virtual screening to find new kinase inhibitors. The study aims to develop a novel hierarchical virtual screening method based on machine learning (ML) and pharmacophore models to find potential JAK1 inhibitors.[12]
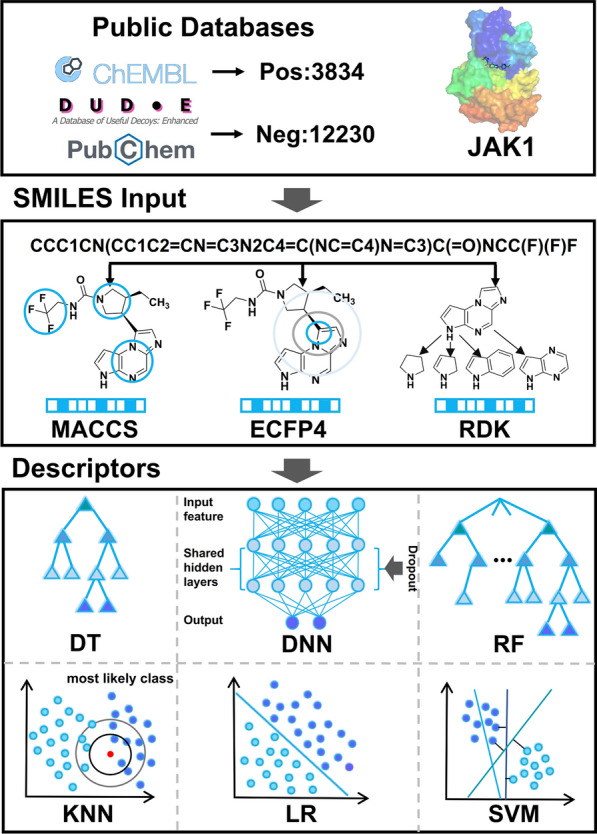
Figure 10. ML Model Building Flowchart
FAQ
Q1: What are the advantages and disadvantages of various experimental methods?
The cost of the ATP residual assay is low. But its conversion rate of at least 50% is required in order to increase signal-to-noise ratio (SNR) greater than two times, so it cannot be used to study kinase properties. The advantage of the method for detecting ADP is that it is highly sensitive and can be applied to a wide range of enzymes without the use of peptide substrates, thus reducing costs. However, it is also susceptible to interference with ATPase activity and cannot measure the activity of kinases in living cells. Fluorescence conjugated energy transfer has a high SNR, stable reading, simple operation, and is convenient for high-throughput quantification, but it is easily affected by proteolytic enzymes, and cannot determine some relevant parameters of peptide substrates, such as the KM value of peptides, nor can some more complex inhibitor mechanism experiments be carried out. Time-resolved fluorescence has strong specificity and a high SNR because the antibody is selective, which avoids the interference of the autofluorescence of the compound itself in the experiment, and the fluorescence signal stabilizes for a longer time without immediate detection. However, specific mAbs are inherently expensive, and not all phosphopeptide substrates are readable. The mobility alteration method has the advantage of allowing real-time monitoring of enzymatic experiments without the addition of abort reagents, but it is difficult to apply in high-throughput screening. The advantage of ligand kinase binding assays is that they can detect low-activity or even inactive protein kinases, or some protein kinases whose biochemical activity is not yet known, and can be easily operated and monitored in real-time. However, the disadvantage is that the specific activity of kinases cannot be obtained, and some compounds that are not inhibited by ATP competition cannot be screened or evaluated.
References
[1] Chan HCS, Shan H, Dahoun T, Vogel H, Yuan S. Advancing Drug Discovery via Artificial Intelligence. Trends Pharmacol Sci. 2019 Aug;40(8):592-604. doi: 10.1016/j.tips.2019.06.004. Epub 2019 Jul 15. Erratum in: Trends Pharmacol Sci. 2019 Oct;40(10):801. PMID: 31320117.
[2] Pinzi L, Rastelli G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int J Mol Sci. 2019 Sep 4;20(18):4331. doi: 10.3390/ijms20184331. PMID: 31487867; PMCID: PMC6769923.
[3] Bedard PL, Hyman DM, Davids MS, Siu LL. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet. 2020 Mar 28;395(10229):1078-1088. doi: 10.1016/S0140-6736(20)30164-1. PMID: 32222192.
[4] Wacker D, Stevens RC, Roth BL. How Ligands Illuminate GPCR Molecular Pharmacology. Cell. 2017 Jul 27;170(3):414-427. doi: 10.1016/j.cell.2017.07.009. PMID: 28753422; PMCID: PMC5560499.
[5] Choi S, Choi KY. Screening-based approaches to identify small molecules that inhibit protein-protein interactions. Expert Opin Drug Discov. 2017 Mar;12(3):293-303. doi: 10.1080/17460441.2017.1280456. Epub 2017 Jan 20. PMID: 28067063.
[6] Goebel GL, Qiu X, Wu P. Kinase-targeting small-molecule inhibitors and emerging bifunctional molecules. Trends Pharmacol Sci. 2022 Oct;43(10):866-881. doi: 10.1016/j.tips.2022.04.006. Epub 2022 May 16. PMID: 35589447.
[7] Lebakken CS, Riddle SM, Singh U, Frazee WJ, Eliason HC, Gao Y, Reichling LJ, Marks BD, Vogel KW. Development and applications of a broad-coverage, TR-FRET-based kinase binding assay platform. J Biomol Screen. 2009 Sep;14(8):924-35. doi: 10.1177/1087057109339207. Epub 2009 Jun 29. PMID: 19564447.
[8] Drueckes P. Protein Kinase Selectivity Profiling Using Microfluid Mobility Shift Assays. Methods Mol Biol. 2016;1439:143-57. doi: 10.1007/978-1-4939-3673-1_9. PMID: 27316993.
[9] He J, Wink S, de Bont H, Le Dévédec S, Zhang Y, van de Water B. FRET biosensor-based kinase inhibitor screen for ERK and AKT activity reveals differential kinase dependencies for proliferation in TNBC cells. Biochem Pharmacol. 2019 Nov;169:113640. doi: 10.1016/j.bcp.2019.113640. Epub 2019 Sep 16. PMID: 31536726.
[10] Gong LL, Fang LH, Peng JH, Liu AL, Du GH. Integration of virtual screening with high-throughput screening for the identification of novel Rho-kinase I inhibitors. J Biotechnol. 2010 Feb 1;145(3):295-303. doi: 10.1016/j.jbiotec.2009.12.003. Epub 2009 Dec 4. PMID: 19963024.
[11] Kyriakou E, Schmidt S, Dodd GT, Pfuhlmann K, Simonds SE, Lenhart D, Geerlof A, Schriever SC, De Angelis M, Schramm KW, Plettenburg O, Cowley MA, Tiganis T, Tschöp MH, Pfluger PT, Sattler M, Messias AC. Celastrol Promotes Weight Loss in Diet-Induced Obesity by Inhibiting the Protein Tyrosine Phosphatases PTP1B and TCPTP in the Hypothalamus. J Med Chem. 2018 Dec 27;61(24):11144-11157. doi: 10.1021/acs.jmedchem.8b01224. Epub 2018 Dec 7. PMID: 30525586.
[12] Wang Z, Sun L, Xu Y, Liang P, Xu K, Huang J. Discovery of novel JAK1 inhibitors through combining machine learning, structure-based pharmacophore modeling and bio-evaluation. J Transl Med. 2023 Aug 28;21(1):579. doi: 10.1186/s12967-023-04443-6. PMID: 37641144; PMCID: PMC10464202.
How to order?

