





Thermal Proteome Profiling (TPP) Service
Thermal proteome profiling (TPP) is a combination of cellular thermal shift assay (CETSA) and mass spectrometry (MS), also termed as MS-CETSA. TPP determines the stability of the entire proteome by measuring the amount of soluble proteins in cells or cell lysates at different heating temperatures. Proteins can change their thermostability when interacting with small molecules (such as drugs or metabolites), nucleic acids, or other proteins, or on post-translational modifications (PTMs), while TPP can identify target proteins based on differences in thermostability with or without ligand-binding.
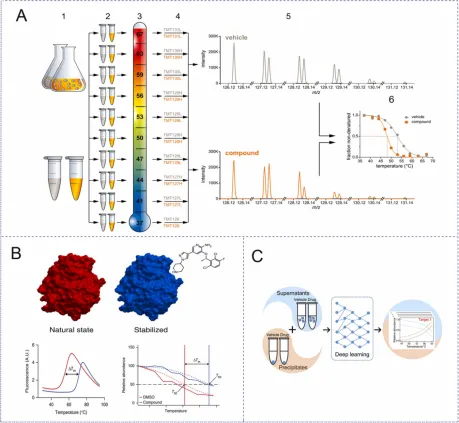
Figure 1. Overview of TPP [1]
1. CETSA
In general, proteins unfold, denature, and precipitate as the heating temperature increases. However, when the ligand binds to its target protein, the thermal stability of the protein can be improved, so that the protein is less susceptible to denaturation and aggregation at the same temperature compared to the unbound protein. The reason for this phenomenon is that ligand-protein complex has a lower energy state than that of native proteins, while the native protein needs to overcome a higher energy barrier to achieve unfolding. Based on the above principles, thermal displacement assays (TSAs) have been well established and applied to the characterization of drug-protein interactions, drug discovery, and structural biology over the past two decades. However, the biggest limitation of traditional TSAs is that they are only suitable for purified proteins. In 2013, Pär Nordlund and colleagues extended TSAs to cellular form, and first developed a conceptually similar technology, called CETSA, which enables direct evaluation of drug-target interactions in a physiological cellular environment.
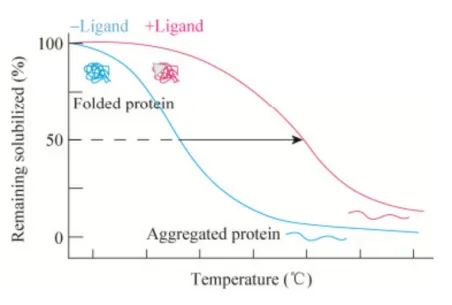
Figure 2. Principles of CETSA
The CETSA workflow consists of the following steps: (1) sample preparation, including live cells, cell lysates or tissue samples; (2) drug processing at specific time; (3) short-time gradient thermal challenge: drug-binding proteins remain relatively stable, while unbound proteins rapidly denature and precipitate at elevated temperatures; (4) cell lysis: intact cells and tissues need to be lysed; (5) cell debris and aggregates are removed from soluble protein fractions by high-speed centrifugation or filtration; (6) residual thermally stable or labile proteins are detected using quantitative techniques such as Western-blotting and MS-based proteomics. Thus, a thermal melting curve (or more precisely, a temperature-induced aggregation curve) of the relevant protein target is established, where the protein Tm is defined as the temperature at which the melting curve value is 0.5. The shift in the melting point curve and the change in Tm value (ΔTm) indirectly reflect the change in the thermal stability of the protein, thus tracking the contact of the drug with the target in the cell. However, it should be noted that the movement of the melting curve can only determine whether there is a drug target interaction at the measured drug concentration, and cannot determine the degree of drug target occupancy (i.e. potency).
The potency of the drug can be determined by isothermal dose-response (ITDR)-CETSA experiment, in which a series of drug concentrations are applied to assess the thermal stability of the drug as a function of dose while the heating temperature remains constant. Thus, ITDR-CETSA can rank the binding ability of a series of compounds to the same target protein by comparing their half-maximum effective dose (EC50). It is important to note that the experimental temperature point is critical to the success of ITDR-CETSA and typically refers to the temperature at which most of the unbound associated proteins are denatured and precipitated in the protein melting curve to ensure that changes in protein thermal stability due to drug binding are clearly observed (typically close to Tm). However, excessive temperatures should be avoided, as this can cause the drug to lose its sensitivity to thermal stability or disrupt the integrity of the cell membrane, which may result in missing weaker binders or erroneous results.
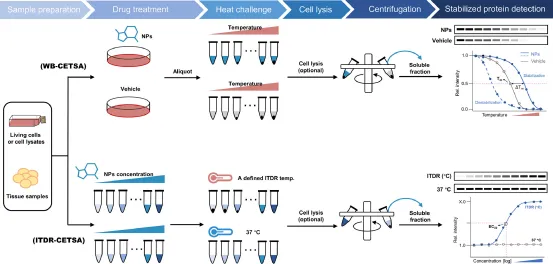
Figure 3. Experimental Procedure for CETSA [2]
CETSA is simple to operate, highly compatible, and can be combined with a variety of readout platforms to form a variety of CETSA variant strategies. Over the past two decades, CETSA and its derived methods have flourished and become powerful tools for all phases of drug discovery and development. According to the method of quantification of the remaining soluble protein after heating, CETSA and its derivatization methods can be divided into three forms.
(1) Classical WB-CETSA: Antibody-based Western blotting analysis was used to detect changes in the thermal stability of the protein of interest. However, this method can only obtain low-throughput results and is limited by the high specificity of antibodies required for WB. This classic CETSA format is often used to validate direct interactions of drug candidates with potential targets identified by other methods in the cellular environment.
(2) TPP, also known as MS-CETSA, that is, the use of MS-based quantitative proteomics or two-dimensional gel electrophoresis (2D-DIGE) for protein identification and quantification, this approach allows for the acquisition of protein stability profiles across the entire proteome in physiologically relevant conditions. TPP and modified TPP methods are ideal for phenotype-based target identification of bioactive compounds in drug discovery.
(3) High-throughput (HT)-CETSA: Convert CETSA to HT format by combining CETSA with homogeneous detection platforms such as AlphaScreen, reporter luminescence, and immunofluorescence. This format enables target-based, large-scale drug discovery in situ by measuring target engagement in a library of compounds at high throughput.
2.TPP
TPP was first launched in 2014, combining the principles of CETSA and multiple quantitative MS introduced the previous year. This method has been widely applied to drug discovery and basic biological research.
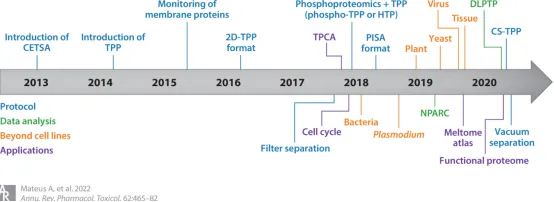
Figure 4. Timeline for the Development and Application of the TPP [3]
TPP typically consists of two types of experiments: a TPP-TR experiment, similar to WB-CETSA, which measures the thermal stability of the entire proteome at a fixed drug concentration, and a TPP-CCR experiment, similar to ITDR-CETSA, which measures the thermal stability of the entire proteome at a fixed temperature point in relation to drug concentration. At the cellular level, the classical TPP-TR process is as follows: cells are first cultured with a saturated concentration of drug or vehicle for a period of time. The treated cells are then divided into 10 aliquots, each of which is heated at different temperature points to induce protein denaturation and aggregation, followed by cell lysis and removal of protein pellets. The remaining soluble protein samples at each temperature were trypsinized and labeled with a multiplex allelic tandem mass tag (TMT10). Then, 10 temperature-treated samples were mixed and imported into LC-MS/MS for large-scale protein identification and quantitation. Protein melting curves and Tm values are indicators to determine whether a protein is a target, and the protein melting curve and Tm value of a drug or vehicle treatment group can be fitted and calculated using specific software.
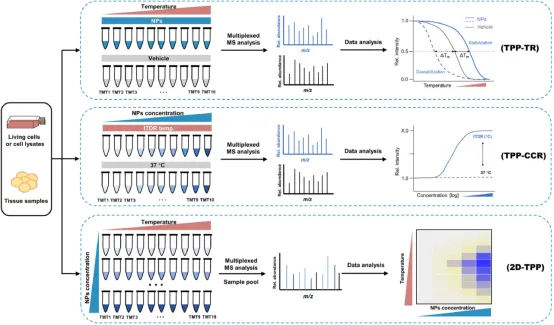
Figure 5. TPP [2]
The TPP-CCR assay is an ideal method for validating the drug targets identified by the TPP-TR assay. In TPP-CCR experiments, cells are treated with increasing concentrations of drug or vehicle controls, and the dose-response curve of the protein and the respective pEC50 values at predetermined temperatures are calculated instead of the melting curve and Tm . In the TPP-TR experiment, even if the thermal stability of the protein is significantly improved under the condition of drug treatment, if the drug is not shown to be dose-dependent in the TPP-CCR experiment, it is likely to be a false positive result. It is important to note that drug-binding does not always increase the thermal stability of the target protein, and drug-induced protein instability has occasionally been observed in TPP experiments. Therefore, it is recommended to perform at least two TPP replicates to eliminate false positive results.
In addition to the classical experimental procedure, the procedure can be modified to obtain new research methods. Two-dimensional thermoproteome analysis (2D-TPP) is a combination of temperature range (where each MS experiment includes all temperatures of a single treatment, i.e., one temperature per channel) and a compound concentration range (where each MS experiment includes multiple compound concentrations at a single temperature, i.e., one compound concentration per channel), which can be a combination of TPP-TR and TPP-CCR, while also combining the advantages of both, and is the most accurate and sensitive current TPP method. The cell material is incubated under N ligands of different concentrations for a certain period of time, and each sample is divided into m aliquots, each sample is heated by one of M temperatures, and the remaining soluble protein is extracted. Proteins are protease-digested and labeled with TMT, and one set of TMT labels can be used for all concentrations and two adjacent temperatures. Perform w=m/2 multiplex MS analysis, identify peptides by database search, and aggregate quantification signals at the protein level. Thus, for each protein I, an M×N data matrix summarizing the reported ionic strength can be obtained, resulting in a heat map colored by the abundance intensity of the soluble protein at each concentration and temperature. In the 2D-TPP method, both the temperature and ligand concentration are systematically varied, which allows for an immediate estimation of the ligand's affinity for the target protein and is more sensitive in identifying the target. Because unlike TPP-TR, all the changing conditions of 2D-TPP are compared in the same mass spectra, resulting in more precise quantification. In addition, in the 2D-TPP method, the relationship between the thermal stability of the target protein and the ligand concentration is expected to be dose-dependent, which adds an additional quality requirement to the data and excludes certain false positives.
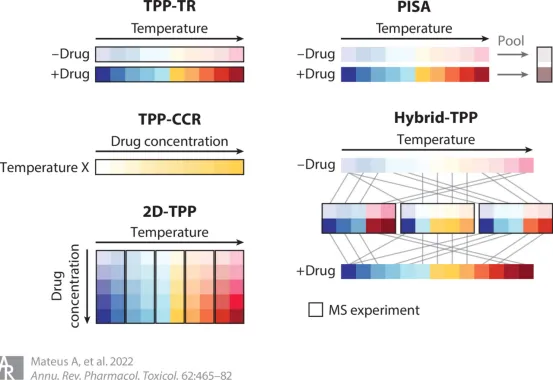
Figure 6. Extension of TPP Method [3]
Proteome Integral Solubility Alteration (PISA) is a recently developed MS-based deep proteomics method for unbiased proteome-wide target deconvolution of ligands without the need for chemical ligand modification. PISA can be applied to live cells to study target engagement in vivo, or to protein extracts to identify proteins for ligand interactions in vitro. The PISA workflow includes ligand incubation using cell cultures or cell lysates, temperature processing, soluble partial separation of the proteome, protein digestion and sample processing for quantitative proteomics, TMT labeling, fractionation, and LC–MS/MS analysis, and data analysis for target deconvolution.
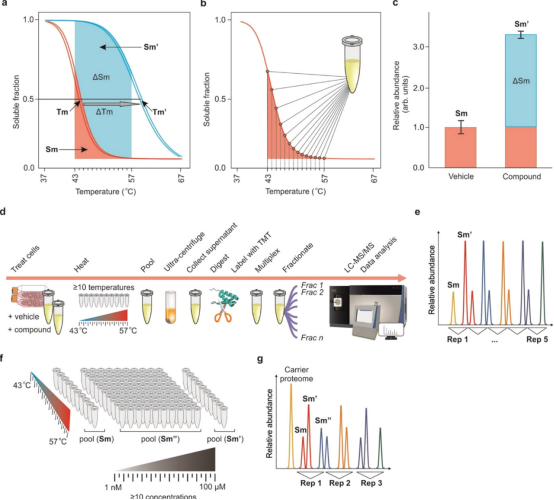
Figure 7. Experimental Process of PISA Method [4]
The TPP method investigated the S-shaped solubility-temperature dependence curve and derived the "melt temperature difference" ΔTm between ligand-treated and vehicle-treated samples. One of the limitations of TPP is the imperfect S-shaped curve, which results in a large number of proteins being discarded. In addition, multiple data points (≥10) need to be collected to construct a single curve, resulting in low experimental throughput. Data collection is limited to a few (often only two) replicates due to the need to control the high cost of analysis, which in turn limits the statistical significance of the results. These problems are addressed in the PISA method, which eliminates the S-shaped solubility-temperature curves and only evaluates the area under these curves. This is achieved by mixing the soluble protein components obtained by processing equal parts of the sample at different temperatures. The protein abundance in this mixed sample is equivalent to the integral of the assumed solubility temperature dependence curve, regardless of its shape. Regardless of the number of temperature points used, the number of samples analyzed by MS in PISA is the same, while in CETSA and TPP, these numbers are linearly dependent. Therefore, PISA can easily detect more temperature points, increase flux, reduce cost and sample size, and/or allow for multiple detection of multiple biological replicates or ligands (such as drugs or compounds of interest) in one experiment. In addition, by removing the curve fitting step that introduces its own uncertainty, PISA provides higher quantitative accuracy for repeated comparisons, thereby improving statistical reliability. To further improve the data quality of PISA, the temperature range can be narrowed down to around the region where the solubility of most proteins varies greatly.
In addition to identifying the direct target of the drug, TPP can also identify indirect targets (i.e., downstream effects caused by initial drug-target binding). TPP performed in cell lysates is thought to reveal direct drug targets due to disruption of signaling protein complexes and dilution of cell contents in lysate systems. However, in living cells, changes in protein thermostability are attributed not only to drug binding, but may also be due to downstream events of drug activation, such as PTMs or binding of physiological ligands (metabolites, nucleic acids, or other proteins). Comparion proteins with altered thermostability identified from cell lysates and intact cells allows for a distinction between direct and indirect targets. Alternatively, shortening the duration of drug treatment is a suggested approach to maximize target occupancy while minimizing the downstream effects of the drug in living cells.
Analysis Workflow
1. Determine the Experimental Process According to the Experimental Requirements
2. Pre-experiment to Optimize Experimental Conditions
3. Cell and Candidate Drug Incubation
4. MS Sample Preparation
5. High Resolution MS Data Collection
6. Data Retrieval and Analysis
Service Advantages
1. Provide Comprehensive Services Such as Optimization of Experimental Conditions and Guidance on Sample Preparation
2. High-Confidence, High-Precision MS Detection
3. Comprehensive Bioinformatics Analysis
Sample Results
1. Identification of OLA1 as a Novel Protein Target of Vitexin by Tissue Thermoproteome Analysis to Ameliorate Dextran Sulfate-Induced Colitis
Vitexin, which is found in a variety of medicinal plants and food sources, has recently gained more attention due to its anti-inflammatory properties. There are studies aimed at identifying vitexin protein targets to ameliorate dextran sodium sulfate (DSS)-induced colitis. The results showed that vitexin not only alleviated the clinical symptoms and colon injury in mice with DSS-induced colitis, but also inhibited the colon production of inflammatory cytokines (IL-1β, IL-6, ICAM and VCAM) and enhanced the expression of barrier-associated proteins (ZO-1, Occludin, and E-cadherin). Based on tissue thermal proteome profiling (Tissue-TPP) and molecular docking, OLA1 was creatively identified as a potential protein target of vitexin. Further siRNA-mediated knockdown of the OLA1 gene in Caco-2 cells demonstrated the ability of OLA1 to increase Nrf2 protein expression and, thus, mediated the anti-inflammatory effects of vitexin. Activation of Nrf2 may require the interaction of the OLA1-vitexin complex with the Keap1 protein to disrupt the Keap1-Nrf2 interaction. The results reveal a novel role of OLA1 as a vitexin protein target, promoting its anti-inflammatory effects by activating Nrf2, which may provide a promising molecular mechanism for novel therapeutic strategies for the treatment of colitis and associated systemic inflammation.
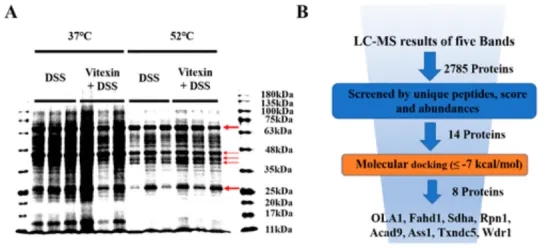
Figure 8. Experimental Process of TPP [5]
2. Label-Free Quantitative TPP Reveals Active Target Transcription Factors Regulated by MC3R Signaling
Label-free quantitative TPP using ion mobility-enhanced liquid chromatography-mass spectrometry (LC-MS) provides a versatile data sets that provides information on protein differential expression, thermal stability, and transcription factor activity. A multidimensional data analysis workflow for label-free quantitative TPP experiments has been developed, which combines genome enrichment analysis, differential protein expression analysis, and inference of transcription factor activity from LC-MS data. It was applied to study downstream signaling processes after activation of the melanocortin 3 receptor (MC3R) by endogenous agonists, which include ACTH, α-MSH, and γ-MSH. The obtained information was used to map signaling pathways downstream of MC3R and to deduce transcription factors responsible for cellular response to ligand treatment. Using this workflow, the study identified the different expressed proteins and investigated their thermal stability. A total of 298 proteins were identified that resulted in altered thermal stability due to MC3R activation. Several of these proteins are transcription factors, suggesting that they are downstream target regulators involved in the MC3R signaling cascade, and altered thermal stability of the transcription factors CCAR2, DDX21, HMGB2, SRSF7, and TET2 was found. These target transcription factors that are evident in the MC3R signaling cascade play an important role in the immune response. In addition, the study inferred the activity of the transcription factors found in the data set. Differential expression data obtained by label-free quantitative LC-MS were used to extrapolate by Bayesian statistics. The inferred transcription factor activity was validated by phosphorylated peptide abundance, highlighting the importance of PTMs in transcription factor regulation.
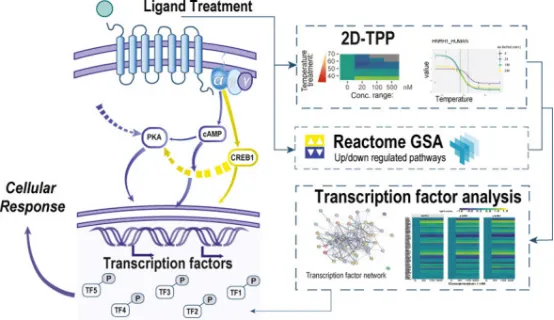
Figure 9. Label-Free Quantitative TPP Reveals Active Target Transcription Factors Regulated by MC3R Signaling [6]
3. Study on Cellular Responses to HSP90 Inhibition in Human Cells Through TPP
Heat shock proteins are chaperones, and they are responsible for protein folding in cells. Heat shock protein 90 (HSP90) is one of the most important chaperones in human cells, and its inhibitory effect is promising in cancer treatment. However, despite the development of several HSP90 inhibitors, none of them are approved for disease treatment due to unexpected cytotoxicity and side effects. Therefore, a more comprehensive study of cellular responses to HSP90 inhibitors can help to better understand the molecular mechanisms of cytotoxicity and side effects of these inhibitors. Changes in the thermal stability of proteins represent alterations in protein structure and interactions and can provide valuable information for the results obtained from commonly used abundance-based proteomic analysis. Cellular responses to different HSP90 inhibitors have been systematically studied using TPP to quantify changes in protein thermal stability globally, as well as measurements of changes in protein abundance. In addition to the target and potential off-target of the drug, proteins with significant changes in thermal stability under HSP90 inhibition are also involved in cellular stress response and translation processes. In addition, proteins with changes in thermostability upon inhibition are located upstream of proteins with altered expression. These findings suggest that HSP90 inhibition disrupts cellular transcription and translation processes. The current research provides a different perspective for a better understanding of cellular responses to chaperone inhibition.
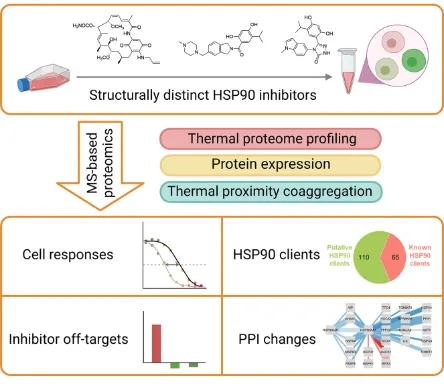
Figure 10. To Study the Cellular Response to HSP90 Inhibition in Human Cells by TPP [7]
Sample Submission Requirements
1. Samples such as drugs/proteins should not be contaminated with impurities as much as possible.
Services at MtoZ Biolabs
1. The Complete Experimental Procedure
2. Relevant Instrument Parameters
3. Raw MS Data
4. Data Analysis Reports
Applications
1.Development and Comparison of New Methods
Target deconvolution is a crucial but costly and time-consuming task, impeding large-scale analysis in drug discovery. A study introduced a matrix-enhanced pooling strategy (MAPS), which mixes multiple drugs in samples with optimized arrangements and uses mathematical processing to simultaneously delineate each drug's targets. We verified this strategy by conducting TPP tests on 15 drugs simultaneously, increasing experimental throughput by 60 times while maintaining high sensitivity and specificity.
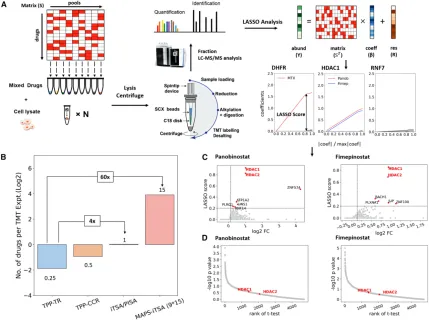
Figure 11. MAPS Workflow and Comparison with Other Methods [8]
2.Data Method Development
Proteins are important macromolecules that perform multiple biological functions. The thermal stability of proteins is a crucial characteristics that affects their function and determines their suitability for various applications. However, current experimental methods, mainly TPP, are expensive, labor-intensive, and limited in proteome and species coverage. To bridge the gap between existing experimental data and sequence information, a study developed a new protein thermal stability predictor called DeepSTABp. DeepSTABp uses a transformer-based protein language model for sequence embedding and state-of-the-art feature extraction, combined with other deep learning techniques for end-to-end protein melting temperature prediction. DeepSTABp can predict the thermal stability of various proteins, making it a powerful and effective tool for large-scale predictions. The model captures structural and biological characteristics that affect protein stability and allows identification of structural features that contribute to protein stability. DeepSTABp is accessible to researchers from all fields through a user-friendly web interface.
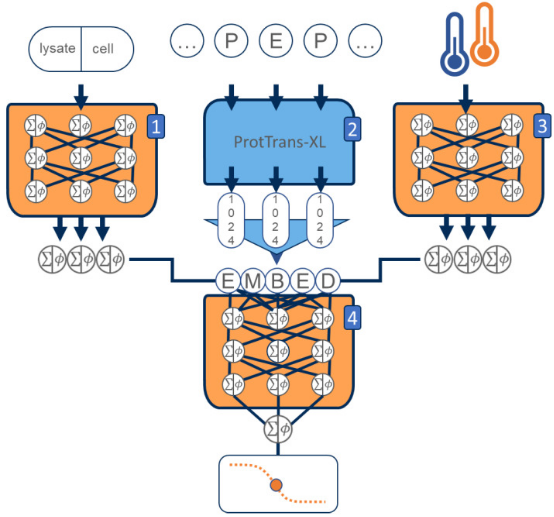
Figure 12. Schematic of the DeepSTABp Deep Learning Model for Predicting Protein Melting Temperatures [9]
3.Drug Target Research
The efficacy of drugs mainly depends on their target binding, while adverse reactions are often caused by toxic off-target binding. From initial discovery to preclinical and clinical research, drug target involvement should be monitored and controlled. CETSA, invented in 2013, provides a powerful tool for measuring direct cellular target engagement in physiological environments based on the principle of ligand-induced target protein thermal stabilization. After nearly a decade of development and optimization, the CETSA strategy has shown universal applicability in various model organisms and has made substantial progress at various stages of drug discovery and development, including WB-CETSA, TPP, and HT-CETSA.
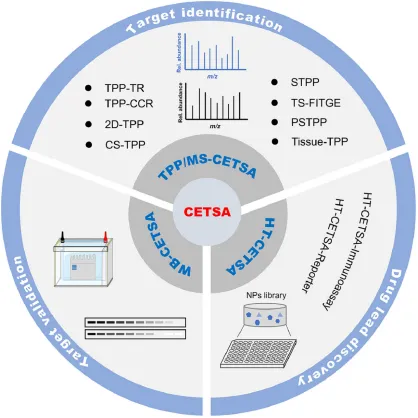
Figure 13. CETSA Strategy Applications in Drug Development [2]
FAQ
Q1: What are the advantages and disadvantages of CETSA and TPP technologies?
CETSA, derived from the traditional TSAs, expands the sample scope from purified proteins to whole cell lysates, complete cells, and even tissues. Initially, the technique only considered soluble proteins, but previous research has demonstrated that membrane-integrated proteins can detect ligand binding through thermal stability. However, CETSA's heating process might affect cell membrane permeability, leading to false positives by allowing drugs that cannot enter cells at physiological temperatures to enter cells. Thus, conditions need to be optimized using the shortest and most effective heating methods, including control experiments to test changes in cell membrane permeability at various temperatures. Additionally, the pre-incubation time of drugs with cells might affect the drug's targeting of specific proteins. If drugs are involved in rapid response signaling pathways, they might cause post-translational changes in the target protein, thereby affecting the antibody's ability to detect the protein when using immunological methods. In such cases, it is advisable to select antibodies that can distinguish between different protein forms or to use MS for target protein identification. If the incubation time is too long, drugs might affect transcription and translation activity, causing protein level changes that affect subsequent detection. Therefore, it is important to control incubation time, typically recommended for 30–60 minutes. In the application of TPP, some proteins' melting curves cannot be reproduced, showing phenomena such as unfolding at low temperatures or only partial melting at high temperatures. The advent of technologies like 2D-TPP allows the use of higher drug concentrations to estimate drug affinity and avoids the measurement of melting curves. Moreover, the emergence of these technologies greatly enhances the sensitivity and rigor of TPP experiments.
References
[1] Feng F, Zhang W, Chai Y, Guo D, Chen X. Label-free target protein characterization for small molecule drugs: recent advances in methods and applications. J Pharm Biomed Anal. 2023 Jan 20;223:115107. doi: 10.1016/j.jpba.2022.115107. Epub 2022 Oct 19. PMID: 36334421.
[2] Tu Y, Tan L, Tao H, Li Y, Liu H. CETSA and profiling strategies for target identification and drug discovery of natural products. Phytomedicine. 2023 Jul 25;116:154862. doi: 10.1016/j.phymed.2023.154862. Epub 2023 May 20. PMID: 37216761.
[3] Mateus A, Kurzawa N, Perrin J, Bergamini G, Savitski MM. Drug Target Identification in Tissues by thermal Proteome Profiling. Annu Rev Pharmacol Toxicol. 2022 Jan 6;62:465-482. doi: 10.1146/annurev-pharmtox-052120-013205. Epub 2021 Sep 9. PMID: 34499524.
[4] Gaetani M, Sabatier P, Saei AA, Beusch CM, Yang Z, Lundström SL, Zubarev RA. Proteome Integral Solubility Alteration: A High-Throughput Proteomics Assay for Target Deconvolution. J Proteome Res. 2019 Nov 1;18(11):4027-4037. doi: 10.1021/acs.jproteome.9b00500. Epub 2019 Oct 14. PMID: 31545609.
[5] Li N, Wang R, Li W, Du Q, Deng Z, Fan Y, Zheng L. Identification of OLA1 as a Novel Protein Target of Vitexin to Ameliorate Dextran Sulfate Sodium-Induced Colitis with Tissue Thermal Proteome Profiling. J Agric Food Chem. 2023 Nov 1;71(43):16057-16066. doi: 10.1021/acs.jafc.3c01559. Epub 2023 Oct 19. PMID: 37856434.
[6] Sandbaumhüter FA, Nezhyva M, Andrén PE, Jansson ET. Label-Free Quantitative Thermal Proteome Profiling Reveals Target Transcription Factors with Activities Modulated by MC3R Signaling. Anal Chem. 2023 Oct 17;95(41):15400-15408. doi: 10.1021/acs.analchem.3c03643. Epub 2023 Oct 7. PMID: 37804223; PMCID: PMC10585664.
[7] Yin K, Wu R. Investigation of Cellular Response to the HSP90 Inhibition in Human Cells Through Thermal Proteome Profiling. Mol Cell Proteomics. 2023 Jun;22(6):100560. doi: 10.1016/j.mcpro.2023.100560. Epub 2023 Apr 27. PMID: 37119972; PMCID: PMC10220490.
[8] Ji H, Lu X, Zhao S, Wang Q, Liao B, Bauer LG, Huber KVM, Luo R, Tian R, Tan CSH. Target deconvolution with matrix-augmented pooling strategy reveals cell-specific drug-protein interactions. Cell Chem Biol. 2023 Aug 28:S2451-9456(23)00274-X. doi: 10.1016/j.chembiol.2023.08.002. Epub ahead of print. PMID: 37652024.
[9] Jung F, Frey K, Zimmer D, Mühlhaus T. DeepSTABp: A Deep Learning Approach for the Prediction of Thermal Protein Stability. Int J Mol Sci. 2023 Apr 18;24(8):7444. doi: 10.3390/ijms24087444. PMID: 37108605; PMCID: PMC10138888.
How to order?

