





Protein Mass Spectrometry Identification Service
Mass spectrometry (MS), a cornerstone tool in modern proteomics research, boasts unparalleled high-throughput analytical capabilities. In the MS-guided protein identification workflow, complex protein samples are first digested into peptides, which are then subjected to a series of intricate manipulations, including efficient separation, precise fragmentation, and ionization, to render them detectable by the mass spectrometer. With its exceptional sensitivity, the mass spectrometer captures and records the characteristic spectra of these peptide ions.
In deciphering these spectra to identify proteins, two primary strategies are employed: database-driven methods and de novo sequencing approaches. Database-driven methods rely on pre-established protein sequence databases, generating theoretical mass spectra patterns through algorithms, which are then meticulously compared with experimentally acquired data to identify the best matches, thereby inferring the protein composition. In contrast, de novo sequencing approaches forgo the need for databases, focusing instead on direct extraction of peptide ion structural information from mass spectra data. Through intricate computational analysis, these approaches gradually construct potential protein sequences, showcasing the sophistication of algorithm design and the ability to deeply mine mass spectra data, thereby pioneering a novel path for protein identification.
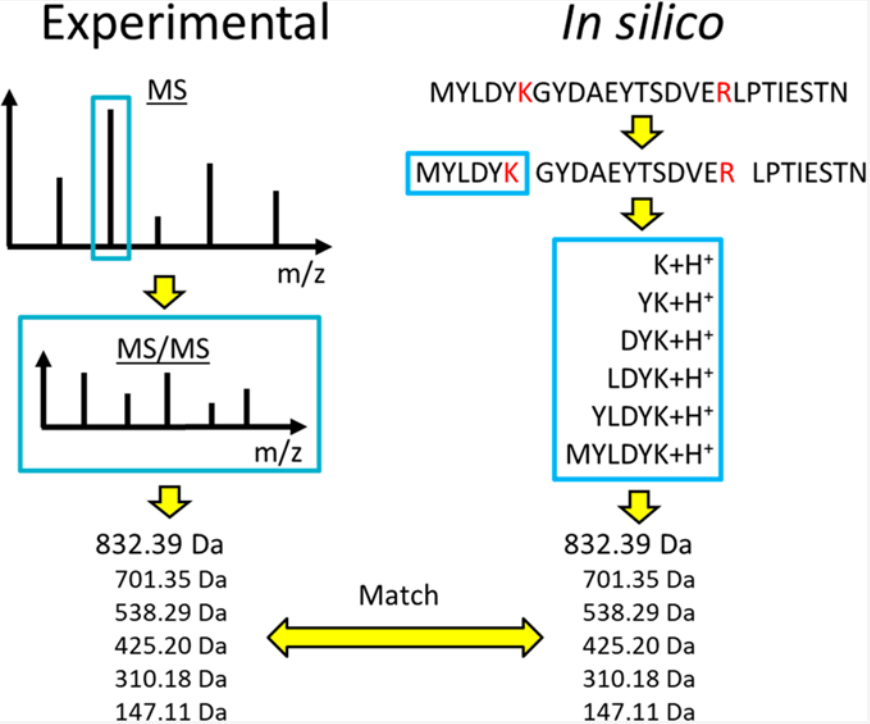
Figure 1. Protein Identification by Database Searching of Mass Spectrometry Data
Service at MtoZ Biolabs
MtoZ Biolabs, a pioneer in protein mass spectrometry analysis, leverages advanced MS platforms and a dedicated service team to offer efficient and precise protein identification services to the global scientific community. We are committed to facilitating groundbreaking research with optimal quality and swift turnaround times. If you have any inquiries or requirements regarding our services, please do not hesitate to contact us to create a new chapter in our scientific collaboration.
Service Advantages
1. Our technology innovatively incorporates a forefront de novo sequencing algorithmic framework, leveraging an intricate data parsing engine to achieve profound analysis and reconstruction of peptide chain sequences, particularly demonstrating unparalleled precision in predicting long sequence tags.
2. We have forsaken the traditional direct matching approach reliant on databases, embracing an innovative intelligent optimization algorithm that effectively handles various uncertainties and errors encountered during sequencing, thereby exhibiting superior robustness and precision.
3. Regarding the scoring mechanism, we have crafted a novel and intricate evaluation model that not only acknowledges the unique merits of de novo sequencing but also integrates multidimensional data considerations, resulting in scoring outcomes that are both scientifically rigorous and logically sound. Compared to alternative methods, our technology markedly enhances protein identification efficiency and diversity while maintaining rigorous error rate control.
Application
1. Protein Modification Analysis
Mass spectrometry enables the identification and quantification of post-translational modifications (PTMs) of proteins, such as phosphorylation, glycosylation, and methylation. These modifications are crucial for protein function and regulation, and mass spectrometry provides precise localization and qualitative means.
2. Protein Structure Analysis
Mass spectrometry can obtain the primary structural information of proteins or peptides, further inferring the secondary and tertiary structural characteristics of proteins. In particular, cross-linking mass spectrometry (XL-MS) technology can provide information on interaction sites within protein complexes, contributing to the understanding of protein spatial structures and functional relationships.
3. Protein Interaction Analysis
Mass spectrometry can be used to study protein-protein, protein-DNA, protein-RNA, and protein-small molecule interactions. Through mass spectrometry analysis after co-immunoprecipitation (Co-IP), the components of protein complexes can be identified, providing critical information for studying protein functions and signaling pathways.
Deliverables
1. Experimental Procedures
2. Relevant Mass Spectrometry Parameters
3. Detailed Information on Protein Mass Spectrometry Identification
4. Mass Spectrometry Images
5. Raw Data
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

