





Ubiquitination Quantitative Proteomic Analysis Service
Ubiquitin (Ub), a small molecule protein composed of 76 amino acids with a molecular weight of approximately 8.5 kDa, is highly conserved throughout evolution. It contains seven lysine (Lys) sites (K6, K11, K27, K29, K33, K48, and K63), one methionine (Met) site at the N-terminus (M1), and one glycine (Gly) site at the C-terminus (G76). Ubiquitin molecules form ubiquitin chains through these sites; if ubiquitin is connected in the same manner, it results in homotypic ubiquitin chains, otherwise, heterotypic ubiquitin chains. Their structure can be linear or branched. Additionally, ubiquitin itself can undergo various modifications. The ubiquitination process typically requires the cooperative action of three ubiquitinating enzymes: E1 ubiquitin-activating enzyme (E1), E2 ubiquitin-conjugating enzyme (E2), and E3 ubiquitin-ligase (E3). In the presence of ATP, E1 activates the ubiquitin molecule and transfers the activated ubiquitin to the E2 enzyme. E3 attaches the ubiquitin binding to E2 to the target protein.
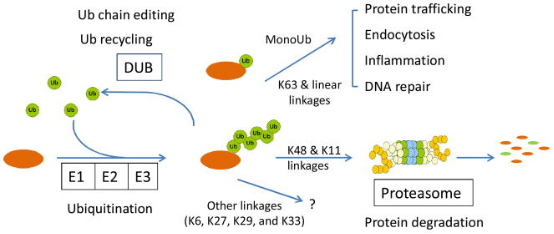
Figure 1. Chemical Properties and Functions of Protein Ubiquitination [3]
Ub and ubiquitin-like proteins (Ubls) are key regulatory factors in cellular events, and dysregulation of the Ub and Ubls exerts influence on the pathogenesis of human diseases. However, the molecular mechanisms of pathogenesis are often unclear, partly due to limitations in analytical tools. The advancement of mass spectrometry (MS) has enabled the biochemical characterization of proteins, providing unprecedented opportunities to dissect the ubiquitin pathway.
Ubiquitination can be categorized into canonical ubiquitination, non-canonical ubiquitination, and free or unanchored polyubiquitin chains. Classical ubiquitination involves the initial ubiquitin being covalently attached to the ε-NH2 group of an internal lysine residue on the target protein. A single ubiquitin modification is defined as monoubiquitination, while Ub modifications on two or more accessible lysine residues on the substrate are referred to as multi-ubiquitination. In most cases, the initial ubiquitin, immediately proximal to the substrate, provides seven lysine sites for potential attachment, resulting in a multi-ubiquitination chain defined by the linkage sites (K6/K11/K27/K29/K33/K48 or K63). Non-canonical ubiquitination occurs on amino acids other than lysine, including methionine, cysteine, serine, or threonine, as well as free amines at the protein N-terminus. Free ubiquitin chains appear in mammals when the UBB and UBC genes express ubiquitin proteins. Unanchored poly-Ub can also be assembled de novo both in vitro and in vivo through specific E2/E3 pairs. Ubiquitin itself undergoes post-translational modifications, such as phosphorylation on 11 sites (T7, T9, T12, T14, S20, T22, T55, S57, Y59, S65, and T66), and acetylation on six lysine residues, except K29.
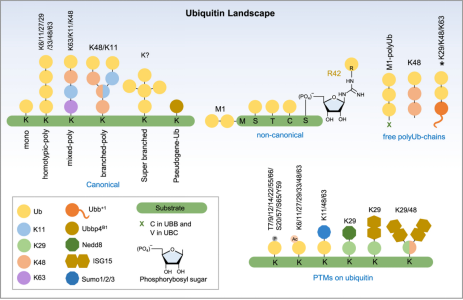
Figure 2. Types of Protein Ubiquitination [4]
The most common method for identifying ubiquitinated proteins and other post-translationally modified proteins is "bottom-up", where proteins are first digested with trypsin. Then it is analyzed by liquid chromatography (LC) tandem mass spectrometry (MS/MS). Various commercial software packages can assign peptide sequences, including PTMs, to MS/MS sequence scan. Unlike other PTMs, ubiquitin is a high molecular weight (8.6 kDa) polypeptide, nearly larger than the associated tryptic peptide of the protein substrate. However, trypsin digestion also cleaves the protein substrate and the ubiquitin PTM. Fortunately, the amino acid sequence near the c-terminus of ubiquitin (-KGG) leaves only a small di-glycine (GG) "remnant" on the lysine ε-NH2 group of the protein substrate peptides. The substrate-modified peptide (‘diGly peptide’) has a mass shift of 114.1 Da (corresponding to GG) and a missed proteolytic cleavage because trypsin cannot engage and cleave the modified lysine of the protein substrate.
1. Methods of Enriching Ubiquitin Conjugates
(1) Affinity Tagged-Ubiquitin Method: In this approach affinity-tagged ubiquitin is trans-expressed in a cellular system followed by affinity chromatography to enrich the tagged-ubiquitin substrate conjugates. The most commonly used tag is 6xHis, combined with metal affinity chromatography under denatured conditions to isolate His-tagged ubiquitin substrate conjugates. A similar approach utilized AviTAG-Ubiquitin expression followed by in-cellulo biotinylation by trans-expression of BirA (Biotin ligase) enzyme and finally pull-down enrichment of the Biotin-AviTAG-Ub-conjugates on streptavidin beads. However, trans-expression of ubiquitin can induce artificial substrate ubiquitination. Also, the inclusion of tagged ubiquitin trans-genes of different substrates and ubiquitin architectures might significantly affect downstream data analysis. Other limitations of this method may be compromised background trypsin peptides on the final GG peptide enrichment and its limitation to cell culture models.
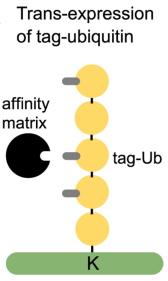
Figure 3. Affinity-Tagged Ubiquitin Method [4]
(2) TUBEs Affinity: Engineered protein binding domains can be used as enrichment reagents for endogenous ubiquitin PTMs. Tandem ubiquitin binding entities (TUBEs) have two or four ubiquitin-binding domains linked together by polyglycine linker, providing nanomolar affinity for polyUb chains. They are typically attached to a tag, such as Halotag, allowing for covalent bead-based enrichment from complex cellular proteomes. By comparing pull-down results with control samples obtained using non-binding mutated TUBE, ubiquitynated proteins can be identified and quantified by MS. The most commonly used TUBE contains the ubiquitin-binding domain UBA of ubiquitin-1, which seems to bind all types of ubiquitin chains. TUBE can also be used to enrich specific types of ubiquitin chains, such as M1/linear (via NEMO UBAN), K29 (via Trabid NZF), K48 (via MINDY-1 tUIM), and K63 (via Tab2 NZF). Non-binding mutated TUBES can be used as negative controls. Overall, TUBE is a powerful tool for enriching ubiquitinated substrates and distinguishing chain types through proteomic techniques. TUBE only enriches polyubiquitin chain conjugates, not free ubiquitin. Additionally, during enrichment, TUBE also protects polyubiquitin chains from DUBs activity and other endogenous ubiquitin binders. One limitation of TUBEs is their limited or non-binding against monoubiquitinated proteins, leaving a gap in proteomic detection of a significant class of ubiquitin PTMs. Furthermore, TUBEs have a tendency to enrich other types of chain except the expected ones.
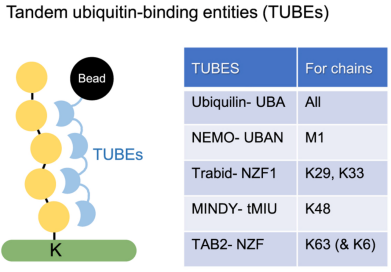
Figure 4. Tandem Ubiquitin Binding Entities (TUBEs) [4]
(3) Through Ubiquitin and Linkage-Specific Antibodies: Antibody-based enrichment of ubiquitin conjugates is a common and widely used method. Linkage-specific antibodies against K11, K27, K29, K48, K63, and M1 have been developed for traditional immunoprecipitation-based workflows. Unlike antibodies, nanobodies consist of a single subunit variable heavy chain (VHH) of ~15–20 kDa and are therefore easily expressed with affinity tags in bacteria to enable high-yield/-purity reagents. Recently, nanobody reagents have been developed with good linkage selectivity for polyubiquitin chains.
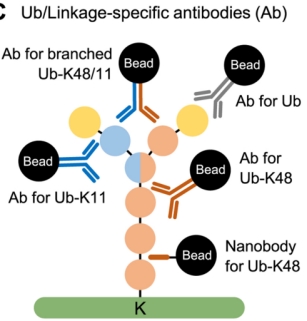
Figure 5. Ubiquitin-specific Antibodies [4]
(4) Linkage-Specific Affimers: As there are no linkage-specific antibodies for K6 and K33 chains, researchers have developed two types of linkage-specific affimers. Affimers are based on the cystatin fold, a 12 kDa non-antibody scaffold, where the randomization of surface loops allows for the generation of large libraries, against which epitopes can be screened and binders can be selected. While these affimers exhibit ~106 fold linkage specificity at the diUb level, when used in a cellular context, appropriate controls must be added to minimize off-target binding at the concentrations typically used in enrichment protocols.
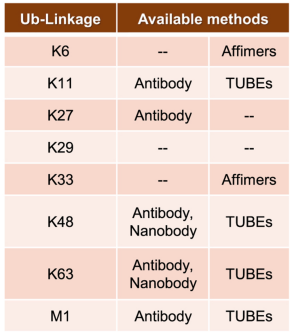
Figure 6. Methods of Enriching Ubiquitination of Different Amino Acids [4]
2. Ubiquitin Peptide Enrichment Methods
(1) By Ub-Remnant Antibodies: A study in 2010 reported a monoclonal antibody for isolating KεGG-modified peptides from the total proteome’s trypsin digests. This reagent anchored the enrichment workflow and enabled the detection of nearly 400 KεGG-modified peptides in mammalian cells via MS. Despite its success, the workflow based on the KεGG antibody indeed has limitations. Firstly, this monoclonal antibody does not recognize GG-modified N-terminal peptides nor ubiquitination sites on non-lysine residues. Secondly, it cannot differentiate between ubiquitination, ISG15, or NEDD8PTM, as they all produce KεGG remnant peptides upon trypsin digestion. To address these issues, a strategy called UbiSite was developed, based on another antibody (UbiSite antibody), which recognizes 13-residue ubiquitin C-terminal remnant (ESTLHLVLRLRGG). After enrichment using UbiSite Ab, these 13-residue modified peptides can be directly subjected to MS/MS identification, or further digested by trypsin to generate GG-modified peptides for subsequent MS/MS analysis. UbiSite workflow can identify N-terminally modified ubiquitination sites. Another monoclonal antibody, called anti-GGX, is used for specific enrichment of complex proteomes. This antibody highly selectively recognizes N-terminal tryptic peptides with a GGM motif and does not cross-react with KεGG remnant peptides.
(2) StUbEx PLUS Method: StUbEx PLUS is an antibody-free high-throughput method to enrich ubiquitination sites at the GG peptide level. In this method of Stable Tagged Ub Exchange (StUbEx) strategy, an endogenous ubiquitin is first knocked down, then stably expressing 6xHis-tagged ubiquitin, resulting in a stable cell line (U2OS). Unlike the N-terminally tagged versions used for protein-level enrichment, in StUbEx, the 6xHis tag is inserted between S65 and T66 positions of ubiquitin. Thus, ubiquitinated substrates can be enriched under denatured conditions through immobilized metal affinity chromatography (Ni-NTA), thus avoiding sequence biases. This workflow also avoids unwanted co-binders of ubiquitin conjugates, DUB activity, and accidental co-purification of NEDD8- and ISG15-modified substrates. Since the His tag is located downstream of the last lysine (Lys63) in the ubiquitin sequence, digestion with LysC on the bead-bound substrates can release all substrate peptides except for the His tag. Subsequent trypsin digestion on the resin produces a highly enriched pool of GG peptides, used for mass spectrometric identification of ubiquitination sites. The limitations of this method include the challenge of generating a stable cell line with labeled ubiquitin due to the presence of four endogenous ubiquitin genes in cells; the position of the 6xHis tag in the ubiquitin tail may cause artificial positive or negative modifications; and the final enrichment rate of GG peptides is affected by the presence of background tryptic peptides (33%).
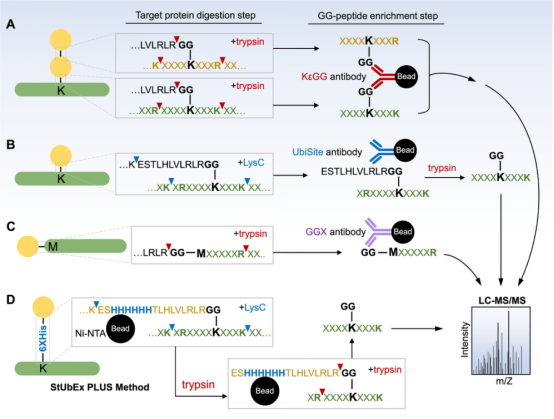
Figure 7. Methods of Enrichment for Ubiquitin Peptides [4]
(3) COFRADIC Method: As an alternative to biochemical methods or trans-gene expression methods, a study developed an antibody-free enrichment workflow known as Ub COmbined FRActional DIagonal Chromatography (COFRADIC). In this method, all free amines in the cell protein extracts (including ubiquitin conjugates) are chemically acetylated. Subsequently, USP2cc is added to remove ubiquitin, reverting the previously modified lysines back to free amines. Then, these newly formed amines are chemically modified with Boc glycine, followed by trypsin digestion. An aliquot analyzed by LC-MS provide the retention times for all peptides, including those previously ubiquitinated with Boc glycine modifications. The remaining samples are treated with trans fatty acid (TFA) to remove the hydrophobic Boc modifications, and then subjected to a second analysis of LC-MS/MS. Compared to the first round of LC-MS analysis, the peptide features detected in MS1 exhibit a significant retention time shift, necessitating MS/MS analysis to determine the sequence and ubiquitin sites identification. The described COFRADIC technology offers an unbiased and effective strategy for enriching ubiquitinated peptides and accurately mapping modified sites. Additionally, the flexibility of this technology lies in the use of DUB - USP2cc; by substituting it with a linkage-specific DUB, it is possible to isolate peptides of interest and identify specific sites. Improvising COFRADIC strategy with deSUMOylases, deNeddylating enzyme (NEDP1), or deISGylating enzymes (USP18, SNEP1/2/3) as alternatives for USP2 the Ubl modification sites can be potentially identified. One potential limitation is the difficulty in achieving stoichiometric acetylation of free amines on lysine residues, which may increase the false positive rate in ubiquitin site identification.
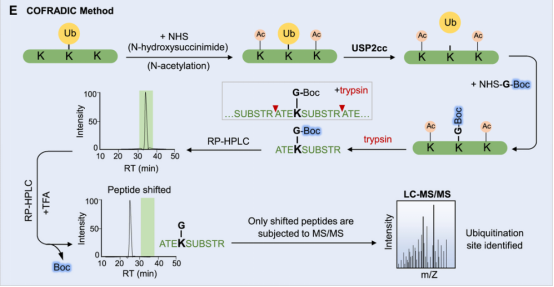
Figure 8. COFRADIC Method [4]
3. Quantitative Strategies for Ubiquitination
(1) Relative Quantification: Relative quantification strategies include two main methods, label-based and label-free. Regardless of the specific method, they are compared to a baseline or control condition. Relative quantification monitors changes across one or more biological states. Labeled-based methods involve metabolic labeling at the protein level, such as Stable Isotope of labeled amino acids in Cell culture (SILAC), or chemical tagging after trypsin digestion of cellular proteins. Commercial kits containing SILAC or chemical agents (iTRAQ or TMT) are widely used. The second major method of relative quantification is "label-free," where peptide mass spectral peak intensities or extracted "native" ion chromatogram areas are directly compared to determine the relative quantification values for proteins or PTMs under different biological conditions. In general, compared to label-based methods, the sample processing workflow for label-free analysis is simplified, although the lack of multiplexing means that each sample must be analyzed separately by LC-MS/MS. Furthermore, label-free methods are applicable to a broader range of mass spectrometers, including low and medium resolution devices. In contrast, label-based methods are better suited for high-resolution devices; for the 10-plex and greater TMT reagents, only high-resolution mass spectrometers can quantify each isotopologue channel.
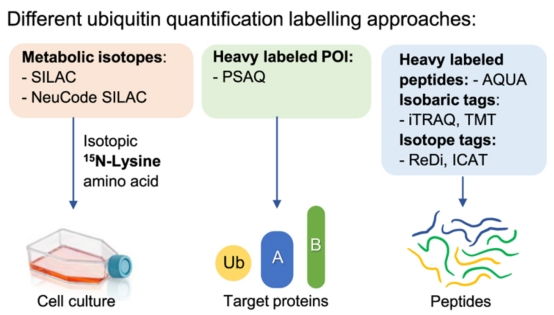
Figure 9. Relative Quantification of Ubiquitination Levels by Labeling Method [4]
(2) Absolute Quantification: One major application of relative ubiquitination quantification is to perform systematic proteome-scale discovery of ubiquitinated proteins and their dynamics relative to control conditions. Once substrates and ubiquitination sites are identified, internal standards can be combined with targeted proteomics methods to obtain stoichiometric information on ubiquitination events in response to various cellular stimuli. Two main targeted methods are widely used for absolute quantification of the ubiquitination landscape—Ubiquitin-Absolute Quantification (Ub-AQUA) and Ubiquitin-Protein standard absolute quantification (Ub-PSAQ). For the Ub-AQUA method, a set of 18 isotopically heavy (13C or 15N) synthetic peptides are used as internal standards to generate calibrate curves for absolute quantification. These reference peptides correspond to trypsin-digested ubiquitin peptides at six major loci on seven lysine residues and the N-terminal methionine with or without GG modifications. After adding the standards to trypsin digests, targeted acquisition (typically multiple reaction monitoring (MRM) or more recently parallel reaction monitoring (PRM)) is used to monitor the intensities of reference and experimental peptides by MS/MS. These acquisition methods provide more measurements or data points for each target peptide, generally improving the quantitative dynamic range and detection limit.
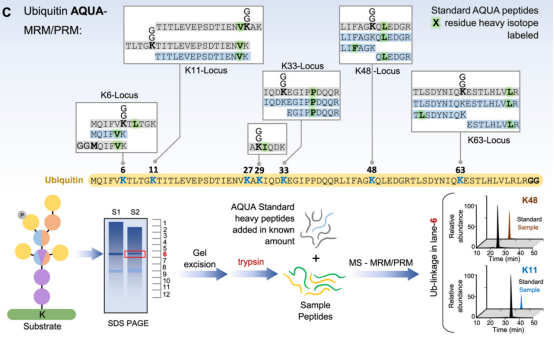
Figure 10. Ubiquitin-AQUA Method Main Process [4]
Ubiquitin-PSAQ method combines differential affinity chromatography and stable isotope labeling (13C or 15N) of free ubiquitin or ubiquitin conjugate standards. This method accurately measures the cellular molar concentrations of free ubiquitin, ubiquitin chains, and monoubiquitinated conjugates in cell extracts. In this strategy, isotope labeled standards (including free Ub, monoUb, and polyUb-conjugates) are spiked into cell or tissue lysates. The mixture of standards and cell ubiquitin conjugates is then divided in half. One half was digested with USP2 followed by selective affinity pull-down using Iso-T zinc-finger (BUZ) domain and for the other half of undigested Ub conjugates hPLIC2-UBA was used for affinity pull-down. Subsequent trypsin digestion and LC-MS/MS analysis allow for quantification of endogenous ubiquitin peptides and GG peptides by comparing the ratio of ion intensities to those of synthetic reference tryptic peptides. The Ub-PSAQ method overcomes several common issues in ubiquitin quantification: (1) tryptic digestion of combined experimental and reference analytes circumvents confounding results due to the addition of peptide standards post-digestion, (2) targeting specific ubiquitin species, (3) utilizing USP2 treated ubiquitin pool for accurate quantification, and (4) avoiding DUB activity by maintaining denatured cell lysis conditions.
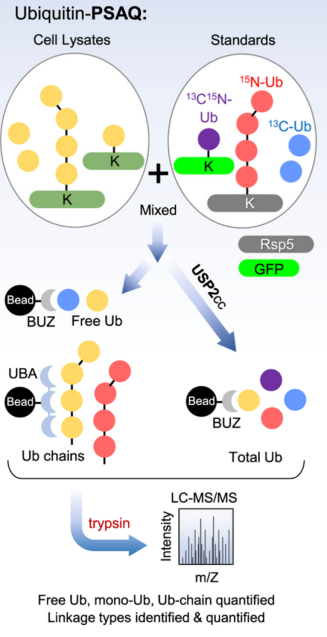
Figure 11. Ubiquitin-PSAQ Method Main Process [4]
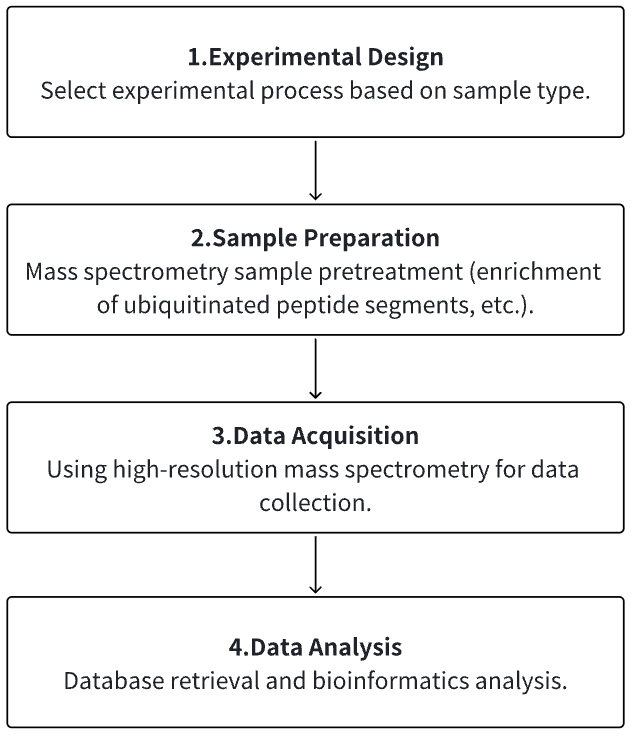
Analysis Workflow
1. Determine the Experimental Process According to the Experimental Requirements
2. Mass Spectrometry Sample Preparation
3. High-resolution Mass Spectrometry Acquisition Data
4. Data Retrieval and Analysis
Service Advantages
1. Identification/Quantification/Modification Identification of Multi-Type Sample Source Proteins
2. High-confidence, High-precision Mass Spectrometry Detection
3. Comprehensive Bioinformatics Analysis
Sample Results
1. Systematic Quantitative Evaluation of the Ubiquitin-Modified Proteome
Although multiple biological pathways are regulated by ubiquitylation, the global identification of ubiquitination target substrates remains a challenge. To globally characterize the human ubiquitin-modifified proteome (ubiquitinome), studies utilized a monoclonal antibody that can recognize diGly-containing isopeptides after trypsin digestion and identified ~19,000 diGly-modified lysine residues in ~5000 proteins. Using quantitative proteomics, the study monitored temporal changes in diGly site abundance in response to proteasomal and translational inhibition, indicating the dependence of observed site abundance changes on ongoing translation and the unique dynamics of individual modified lysine residues in response to proteasomal inhibition. Additionally, it demonstrated that quantitative diGly proteomics can be used to identify substrates of cullin-RING ubiquitin ligases. Characterization of the ubiquitinome not only allowed for the quantitative assessment of changes in protein homeostasis fidelity but also identifies substrates of individual ubiquitin pathway enzymes.
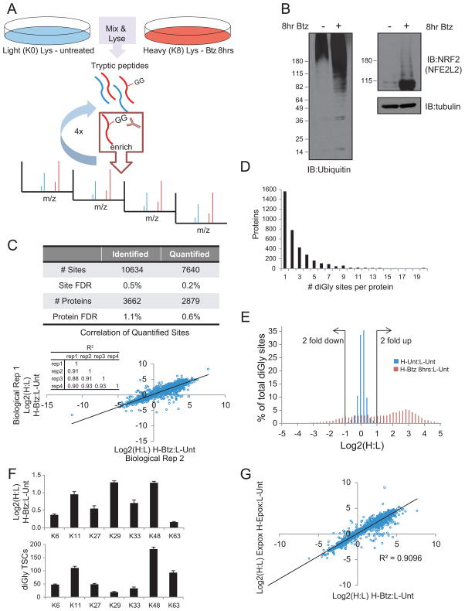
Figure 12. Overview of diGly Proteomics Enrichment Strategy [5]
2. Quantitative Lys-ε-Gly-Gly (diGly) Proteomics Combined with Inducible RNAi Reveals E3 Ligase HUWE1 Mediated Ubiquitin Hydrolysis of DNA Damage-Induced Transcript 4 (DDIT4)
Through the ubiquitin-proteasome system (UPS), the activity of E3 ubiquitin ligases targets proteins for degradation, regulating various cellular processes, and dysregulation of these enzymes contributes to the pathogenesis of human diseases. One of the challenges in the UPS field is to depict the complete cohort of substrates for a particular E3 ligase. Advances in mass spectrometry and the development of antibodies that recognize Lys-ε-Gly-Gly (diGly) remnant trypsinolysis of ubiquitinated proteins provide tools to address this issue. A study implemented an inducible loss-of-function approach, combined with quantitative diGly proteomics, to find new substrates for HUWE1, an E3 ubiquitin protein ligase associated with cancer and intellectual disabilities. The diGly proteomics results led to the identification of DNA damage induced transcript 4 (DDIT4) as a presumed substrate of HUWE1. Cell-based assays showed that HUWE1 interacted with and regulated the ubiquitination and stability of DDIT4, suggesting a model of HUWE1-mediated degradation of DDIT4 proteasomal. The findings provided proof of concept that inducible knockdown of E3 ligases combined with diGly proteomics offered a potentially advantageous approach to identifying new E3 substrates, which may help pinpoint therapeutic targets in the UPS.
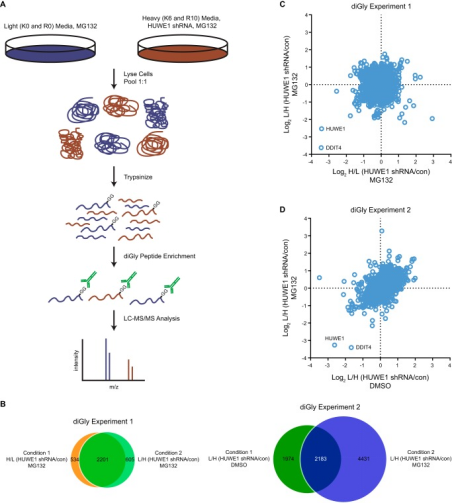
Figure 13. Quantitative diGly Proteomics Workflow [6]
3. Ubiquitin Proteomics Identifies RNA Polymerase I as a Target of the Smc5/6 complex
Ubiquitination controls many cellular processes and its dysregulation is associated with many pathologies. The Nse1 subunit of the Smc5/6 complex contains a RING domain with ubiquitin E3 ligase activity and essential functions in genome integrity. However, Nse1-dependent ubiquitin targets remain elusive. Research using label-free quantitative proteomics analyzed the nuclear ubiquitinome of nse1-C274A RING mutant cells. Results indicated that Nse1 influences the ubiquitination of several proteins involved in ribosome biogenesis and metabolism, importantly, beyond the typical functions of the Smc5/6 complex. Analysis suggested a link between Nse1 and ubiquitination of RNA Polymerase I (RNA Pol I). Specifically, Nse1 and the Smc5/6 complex promoted ubiquitination at K408 and K410 within the Rpa190 clamp domain, a modification that induced its degradation in response to transcription elongation stalling. It is believed that this mechanism contributes to the Smc5/6-dependent disassembly of rDNA arrays, sites transcribed by RNA Pol I.
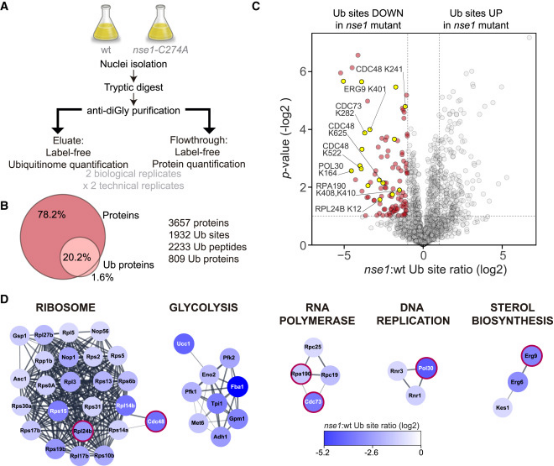
Figure 14. Proteomic Screening to Identify Nse1 Sensitive Ub Sites [7]
4. Quantitative Ubiquitome Analysis Reveals the Specificity of RNF111/Arkadia E3 Ubiquitin Ligase for its Degradation Substrates SKI and SKIL/SnoN in the TGF-β Signaling Pathway
RNF111/Arkadia is an E3 ubiquitin ligase that activates the transforming growth factor-β (TGF-β) pathway by degrading the transcriptional inhibitors SKIL/SnoN and SKI. Studies using integrative proteomics analyzed the endogenous substrates of RNF111 after activation of TGF-β signaling in parental U2OS cell lines, enriched for ubiquitinated proteins (ubiquitinome) and compared with U2OS CRISPR engineered clones expressing a truncated form of RNF111 without its C-terminal RING domain. Prior to proteomic analysis via mass spectrometry, two methods of ubiquitylated protein enrichment were compared: one using K-ε-GG antibodies for diGly remnant peptide immunoprecipitation and another using a new method of protein immunoprecipitation recognizing all ubiquitin chains and mono-ubiquitylation with pan nanobody. The comprehensive comparison of RNF111-dependent proteome and ubiquitylomes could identify SKI and SKIL as the only targets ubiquitinated and degraded by RNF111 E3 ligase in the presence of TGF-β. The lysine 343 in the SAND domain of SKIL constituted the target of RNF111 ubiquitination, demonstrating the specificity of RNF111 E3 ligase function targeting SKI and SKIL for ubiquitination and degradation after TGF-β pathway activation.
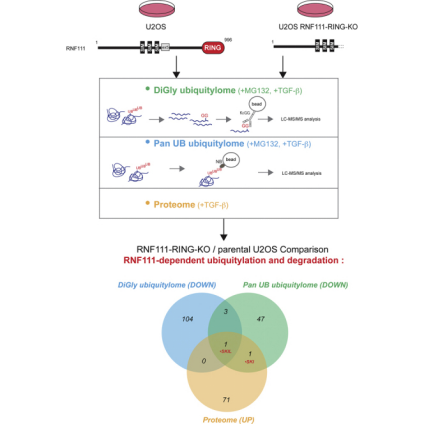
Figure 15. Quantitative Ubiquitinome Study Workflow [8]
Sample Submission Requirements
1. Protein Purity >90%
2. Try to Avoid Impurity Contamination
Services at MtoZ Biolabs
1. Experimental Procedure
2. Relevant Mass Spectrometry Parameters
3. Raw Data
4. Data Analysis Reports
Applications
1. Quantitative Ubiquitinated Proteomics Research Signaling Pathways
Ubiquitin (UB) has become the main signal which controlling flux through signaling pathways, and this modification is often combined with phosphorylation to determine the timing and flow of information in cells. The 76-amino acid UB is linked to lysine residues in target proteins through the E1-E2-E3 cascade, and the ubiquitylation mechanism also promotes the formation of the described UB-UB linkages arrays, which regulate information (degradation, signal transduction, recruitment, etc.) delivered to target proteins. The ubiquitin (UB)-driven signal transduction system permeates biology and is typically integrated with other types of post-translational modifications (PTM), most notably phosphorylation. The flux through these pathways is usually determined by the fractional stoichiometry of different regulatory modifications and protein assemblies, as well as the spatial organization of pathway components. Quantitative proteomic tools and enrichment strategies can be used to quantify UB-dependent signal transduction systems, and can combine UB signal transduction with regulatory phosphorylation events to determine changes in cellular signaling pathways.
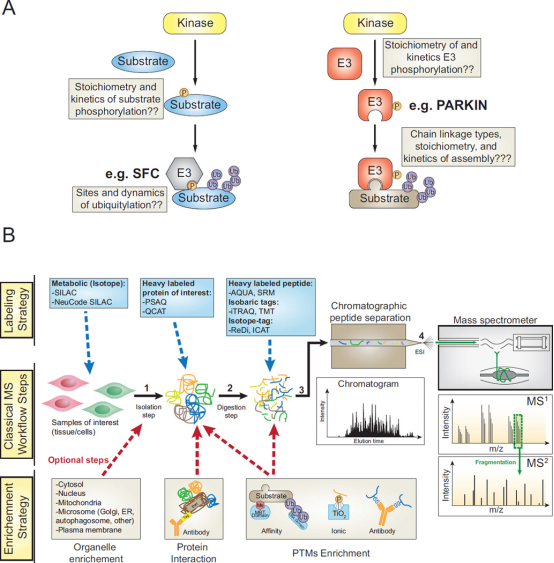
Figure 16. Quantitative Ubiquitinome Study for Signal Transduction [9]
2. Ubiquitin Proteomics Research on Aging
Intracellular unwanted, damaged, or misfolded proteins are tagged with ubiquitin chains. This tag serves as a signal for destruction by the 26S proteasome, an enzyme complex that degrades proteins to clear out them. Studies have shown that in aging nematodes caenorhabditis elegans, some proteins escape the clearance of the ubiquitin-proteasome system, and reduce the accumulation of these proteins can extend life. Research used mass spectrometry to measure levels of different proteins in nematode cells of various ages. Ubiquitinated proteins were quantified by purifying them using antibodies that can recognize signs of previous ubiquitination on protease-digested protein fragments. Compared to young nematodes, ubiquitination in old nematodes was generally reduced.
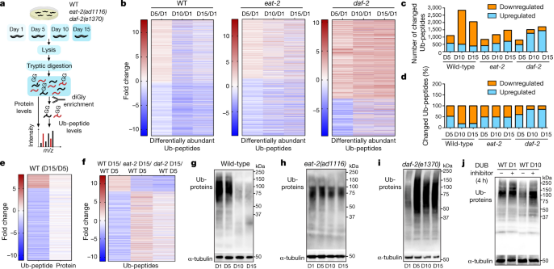
Figure 17. Ubiquitin Proteomics Research on Aging in Caenorhabditis Elegans [10]
FAQ
Q1: Besides quantifying ubiquitination, is it possible to identify the structure of ubiquitin chains?
Common bottom-up approaches involve the digestion steps of protein hydrolysis and folding complex polyubiquitin chains into a common residue, which effectively mask key information about the Ub structure. To circumvent this limitation and preserve some information about the polyubiquitin structure, methods have been developed using chain-specific antibodies, chain-specific DUBs, affinity purifiers, and top-down approaches, along with quantitative strategies.
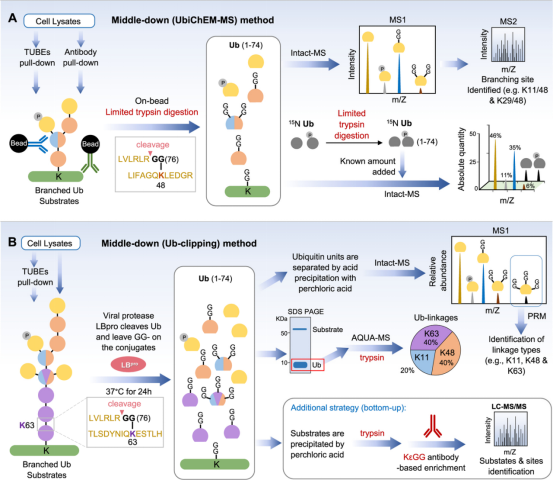
Figure 18. Determination of Ubiquitin Chain Structure [4]
References
[1] Cox J, Mann M. Quantitative, high-resolution proteomics for data-driven systems biology. Annu Rev Biochem. 2011;80:273-99. doi: 10.1146/annurev-biochem-061308-093216. PMID: 21548781.
[2] Ahire D, Kruger L, Sharma S, Mettu VS, Basit A, Prasad B. Quantitative Proteomics in Translational Absorption, Distribution, Metabolism, and Excretion and Precision Medicine. Pharmacol Rev. 2022 Jul;74(3):769-796. doi: 10.1124/pharmrev.121.000449. PMID: 35738681; PMCID: PMC9553121.
[3] Chen PC, Na CH, Peng J. Quantitative proteomics to decipher ubiquitin signaling. Amino Acids. 2012 Sep;43(3):1049-60. doi: 10.1007/s00726-012-1286-y. Epub 2012 Jul 22. PMID: 22821265; PMCID: PMC3498854.
[4] Sahu I, Zhu H, Buhrlage SJ, Marto JA. Proteomic approaches to study ubiquitinomics. Biochim Biophys Acta Gene Regul Mech. 2023 Jun;1866(2):194940. doi: 10.1016/j.bbagrm.2023.194940. Epub 2023 Apr 29. PMID: 37121501; PMCID: PMC10612121.
[5] Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, Harper JW, Gygi SP. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011 Oct 21;44(2):325-40. doi: 10.1016/j.molcel.2011.08.025. Epub 2011 Sep 8. PMID: 21906983; PMCID: PMC3200427.
[6] Thompson JW, Nagel J, Hoving S, Gerrits B, Bauer A, Thomas JR, Kirschner MW, Schirle M, Luchansky SJ. Quantitative Lys-ϵ-Gly-Gly (diGly) proteomics coupled with inducible RNAi reveals ubiquitin-mediated proteolysis of DNA damage-inducible transcript 4 (DDIT4) by the E3 ligase HUWE1. J Biol Chem. 2014 Oct 17;289(42):28942-55. doi: 10.1074/jbc.M114.573352. Epub 2014 Aug 21. PMID: 25147182; PMCID: PMC4200252.
[7] Ibars E, Codina-Fabra J, Bellí G, Casas C, Tarrés M, Solé-Soler R, Lorite NP, Ximénez-Embún P, Muñoz J, Colomina N, Torres-Rosell J. Ubiquitin proteomics identifies RNA polymerase I as a target of the Smc5/6 complex. Cell Rep. 2023 May 30;42(5):112463. doi: 10.1016/j.celrep.2023.112463. Epub 2023 May 3. PMID: 37141096.
[8] Laigle V, Dingli F, Amhaz S, Perron T, Chouchène M, Colasse S, Petit I, Poullet P, Loew D, Prunier C, Levy L. Quantitative Ubiquitylome Analysis Reveals the Specificity of RNF111/Arkadia E3 Ubiquitin Ligase for its Degradative Substrates SKI and SKIL/SnoN in TGF-β Signaling Pathway. Mol Cell Proteomics. 2021;20:100173. doi: 10.1016/j.mcpro.2021.100173. Epub 2021 Nov 3. PMID: 34740826; PMCID: PMC8665411.
[9] Ordureau A, Münch C, Harper JW. Quantifying ubiquitin signaling. Mol Cell. 2015 May 21;58(4):660-76. doi: 10.1016/j.molcel.2015.02.020. PMID: 26000850; PMCID: PMC4441763.
[10] Koyuncu, S., Loureiro, R., Lee, H.J. et al. Rewiring of the ubiquitinated proteome determines ageing in C. elegans. Nature 596, 285–290 (2021). https://doi.org/10.1038/s41586-021-03781-z.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
Post-Translational Modifications Proteomics Service
How to order?

